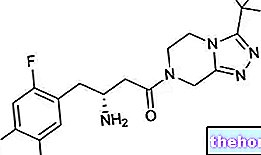
Actieve ingrediënten: Sitagliptine
Xelevia 100 mg filmomhulde tabletten
Xelevia-bijsluiters zijn beschikbaar voor verpakkingsgrootten:- Xelevia 25 mg filmomhulde tabletten
- Xelevia 50 mg filmomhulde tabletten
- Xelevia 100 mg filmomhulde tabletten
Waarom wordt Xelevia gebruikt? Waar is het voor?
Xelevia bevat de werkzame stof sitagliptine die behoort tot een klasse geneesmiddelen die dipeptidylpeptidase-4 (DPP-4)-remmers worden genoemd en die de bloedsuikerspiegel verlagen bij volwassen patiënten met type 2-diabetes mellitus.
Dit geneesmiddel helpt de insulinespiegels die na een maaltijd worden geproduceerd te verhogen en vermindert de hoeveelheid suiker die door het lichaam wordt geproduceerd.
Uw arts heeft dit geneesmiddel voorgeschreven om u te helpen uw bloedsuikerspiegel te verlagen, die te hoog is vanwege diabetes type 2. Dit geneesmiddel kan alleen of samen met andere geneesmiddelen (insuline, metformine, sulfonylureumderivaten of glitazonen) worden gebruikt die de bloedsuikerspiegel verlagen die u misschien al gebruikt om uw diabetes te behandelen, samen met een dieet- en bewegingsprogramma.
Wat is diabetes type 2?
Diabetes type 2 is een ziekte waarbij het lichaam niet genoeg insuline aanmaakt en de door het lichaam aangemaakte insuline niet zo goed werkt als zou moeten. Het lichaam kan ook te veel suiker aanmaken. Wanneer dit gebeurt, hoopt zich suiker (glucose) op in het bloed. Dit kan leiden tot ernstige medische problemen zoals hartaandoeningen, nieraandoeningen, blindheid en amputaties.
Contra-indicaties Wanneer Xelevia niet mag worden gebruikt
Gebruik Xelevia niet
- als u allergisch bent voor sitagliptine of voor één van de andere bestanddelen van dit geneesmiddel.
Voorzorgen bij gebruik Wat u moet weten voordat u Xelevia inneemt
Gevallen van ontsteking van de alvleesklier (pancreatitis) zijn gemeld bij patiënten die met Xelevia werden behandeld.
Vertel het uw arts als u:
- een pancreasziekte (zoals pancreatitis)
- galstenen, alcoholverslaving of zeer hoge niveaus van triglyceriden (een vorm van vet) in het bloed. Deze medische aandoeningen kunnen het risico op het ontwikkelen van pancreatitis vergroten
- diabetes type 1
- diabetische ketoacidose (een complicatie van diabetes met hoge bloedsuikerspiegel, snel gewichtsverlies, misselijkheid of braken)
- eventuele vroegere of huidige nierproblemen
- een allergische reactie op Xelevia.
Het is onwaarschijnlijk dat dit geneesmiddel een lage bloedsuikerspiegel (hypoglykemie) veroorzaakt, omdat het niet werkt als uw bloedsuikerspiegel laag is. Wanneer dit geneesmiddel echter wordt ingenomen met een sulfonylureumderivaat of met insuline, kan (hypoglykemie) optreden. Uw arts kan de dosis van het sulfonylureumderivaat of insuline verlagen.
Kinderen en adolescenten
Kinderen en jongeren onder de 18 jaar mogen dit geneesmiddel niet gebruiken. Het is niet bekend of het gebruik van dit geneesmiddel veilig en effectief is bij kinderen en jongeren onder de 18 jaar.
Interacties Welke medicijnen of voedingsmiddelen kunnen het effect van Xelevia veranderen?
Vertel het uw arts of apotheker als u andere geneesmiddelen gebruikt, kort geleden heeft gebruikt of in de nabije toekomst gaat gebruiken.
Vertel het uw arts in het bijzonder als u digoxine gebruikt (een geneesmiddel dat wordt gebruikt om een onregelmatige hartslag en andere hartproblemen te behandelen). Het digoxinegehalte in uw bloed moet mogelijk worden gecontroleerd als het samen met Xelevia wordt ingenomen.
Waarschuwingen Het is belangrijk om te weten dat:
Zwangerschap en borstvoeding
Als u zwanger bent of borstvoeding geeft, denkt zwanger te zijn of zwanger wilt worden, vraag dan uw arts of apotheker om advies voordat u dit geneesmiddel inneemt. U mag dit geneesmiddel niet gebruiken tijdens de zwangerschap.
Het is niet bekend of dit geneesmiddel in de moedermelk terechtkomt. U mag dit geneesmiddel niet gebruiken als u borstvoeding geeft of denkt dat u borstvoeding moet geven.
Rijvaardigheid en het gebruik van machines
Dit geneesmiddel heeft geen of een verwaarloosbare invloed op de rijvaardigheid en op het vermogen om machines te bedienen. Duizeligheid en slaperigheid zijn echter gemeld, die uw rijvaardigheid en uw vermogen om machines te bedienen kunnen beïnvloeden.
Het gebruik van dit geneesmiddel met andere geneesmiddelen, sulfonylureumderivaten genaamd, of met insuline kan hypoglykemie veroorzaken, die uw rijvaardigheid, het vermogen om machines te bedienen of te werken zonder beschermende barrières kan beïnvloeden.
Dosis, wijze en tijdstip van toediening Hoe wordt Xelevia gebruikt: Dosering
Gebruik dit geneesmiddel altijd precies zoals uw arts u dat heeft verteld. Raadpleeg bij twijfel uw arts of apotheker.
De gebruikelijke aanbevolen dosis is:
- één filmomhulde tablet van 100 mg
- een keer per dag
- mondeling
Als u nierproblemen heeft, kan uw arts lagere doses voorschrijven (zoals 25 mg of 50 mg).
U kunt dit geneesmiddel met of zonder eten en drinken innemen.
Uw arts kan dit geneesmiddel alleen of samen met andere geneesmiddelen voorschrijven die uw bloedsuikerspiegel verlagen.
Dieet en lichaamsbeweging kunnen uw lichaam helpen de bloedsuikerspiegel beter te gebruiken. Het is belangrijk om het door uw arts aanbevolen dieet en trainingsprogramma voort te zetten terwijl u Xelevia gebruikt.
Bent u vergeten Xelevia in te nemen?
Als u een dosis bent vergeten, neem deze dan in zodra u eraan denkt. Als u het zich niet herinnert tot uw volgende dosis moet zijn, sla dan de gemiste dosis over en ga verder met uw normale dosis.
Neem geen dubbele dosis van dit geneesmiddel.
Als u stopt met het innemen van Xelevia
Blijf dit geneesmiddel innemen zolang uw arts het voorschrijft, zodat u uw bloedsuikerspiegel kunt blijven controleren.U mag niet stoppen met het gebruik van dit geneesmiddel zonder eerst met uw arts te overleggen.
Als u nog vragen heeft over het gebruik van dit geneesmiddel, neem dan contact op met uw arts of apotheker.
Overdosering Wat moet u doen als u te veel Xelevia heeft ingenomen?
Als u meer dan de voorgeschreven dosering van dit geneesmiddel heeft ingenomen, neem dan onmiddellijk contact op met uw arts.
Bijwerkingen Wat zijn de bijwerkingen van Xelevia
Zoals alle geneesmiddelen kan ook dit geneesmiddel bijwerkingen hebben, al krijgt niet iedereen daarmee te maken.
STOP met het innemen van Xelevia en neem onmiddellijk contact op met een arts als u een van de volgende ernstige bijwerkingen opmerkt:
- Ernstige en aanhoudende pijn in de buik (maagstreek) die zich naar de rug kan uitstrekken met of zonder misselijkheid en braken, aangezien dit tekenen kunnen zijn van een ontsteking van de alvleesklier (pancreatitis).
Als u een ernstige allergische reactie heeft (frequentie niet bekend), waaronder uitslag, netelroos, blaren op de huid/afschilferende huid en zwelling van het gezicht, de lippen, tong en keel die ademhalings- of slikproblemen kunnen veroorzaken, stop dan met de behandeling met dit geneesmiddel en neem onmiddellijk contact op met uw arts. Uw arts kan u een geneesmiddel voorschrijven om uw allergische reactie te behandelen en een ander geneesmiddel voor uw diabetes.
Sommige patiënten hebben de volgende bijwerkingen gekregen na toevoeging van sitagliptine aan metformine:
- Vaak (komen voor bij minder dan 1 op de 10 gebruikers): lage bloedsuikerspiegel, misselijkheid, winderigheid, braken
- Soms (komen voor bij minder dan 1 op de 100 gebruikers): maagpijn, diarree, constipatie, slaperigheid
Sommige patiënten hebben melding gemaakt van verschillende soorten maagpijn bij het starten van sitagliptine en metformine samen als onderdeel van de combinatietherapie (frequentie komt vaak voor).
Sommige patiënten hebben de volgende bijwerkingen ondervonden bij gebruik van sitagliptine in combinatie met een sulfonylureumderivaat en metformine:
- Zeer vaak (komen voor bij meer dan 1 op de 10 gebruikers): lage bloedsuiker
- Vaak: constipatie
Sommige patiënten hebben de volgende bijwerkingen ondervonden bij het gebruik van sitagliptine en pioglitazon:
- Vaak: winderigheid, zwelling van de handen of benen
Sommige patiënten hebben de volgende bijwerkingen ondervonden bij gebruik van sitagliptine in combinatie met pioglitazon en metformine:
- Vaak: zwelling van de handen of benen
Sommige patiënten hebben de volgende bijwerkingen gekregen bij gebruik van sitagliptine in combinatie met insuline (met of zonder metformine):
- Vaak: griep
- Soms: droge mond
Sommige patiënten hebben de volgende bijwerkingen ondervonden bij gebruik van alleen sitagliptine in klinische onderzoeken, of tijdens gebruik na goedkeuring alleen en/of in combinatie met andere diabetesgeneesmiddelen:
- Vaak: lage bloedsuikerspiegel, hoofdpijn, infectie van de bovenste luchtwegen, loopneus of verstopte neus en keelpijn, artrose, pijn in de armen of benen
- Soms: duizeligheid, constipatie, jeuk
- Frequentie niet bekend: nierproblemen (waarvoor soms dialyse nodig is), braken, gewrichtspijn, spierpijn, rugpijn, interstitiële longziekte
Melding van bijwerkingen
Krijgt u last van bijwerkingen, neem dan contact op met uw arts, apotheker of verpleegkundige.Dit geldt ook voor mogelijke bijwerkingen die niet in deze bijsluiter staan.U kunt bijwerkingen ook rechtstreeks melden via het nationale meldsysteem zoals vermeld in aanhangsel V. meer informatie geven over de veiligheid van dit geneesmiddel.
Vervaldatum en retentie
Buiten het zicht en bereik van kinderen houden.
Gebruik dit geneesmiddel niet meer na de uiterste houdbaarheidsdatum. Die is te vinden op de blisterverpakking en de doos na "EXP". De vervaldatum verwijst naar de laatste dag van die maand.
Voor dit geneesmiddel zijn er geen speciale bewaarcondities.
Gooi geneesmiddelen niet weg via het afvalwater of met huishoudelijk afval. Vraag uw apotheker wat u met geneesmiddelen moet doen die u niet meer gebruikt. Dit helpt het milieu te beschermen.
Andere informatie
Wat bevat Xelevia
- Het werkzame bestanddeel is sitagliptine. Elke filmomhulde tablet (tablet) bevat sitagliptinefosfaatmonohydraat, overeenkomend met 100 mg sitagliptine.
- De andere stoffen in dit middel zijn: in de tabletkern: microkristallijne cellulose (E460), watervrij calciumwaterstoffosfaat (E341), croscarmellosenatrium (E468), magnesiumstearaat (E470b) en natriumstearylfumaraat. De tabletomhulling bevat: poly (vinylalcohol), macrogol 3350, talk (E553b), titaniumdioxide (E171), rood ijzeroxide (E172) en geel ijzeroxide (E172).
Hoe ziet Xelevia eruit en wat is de inhoud van de verpakking
Ronde, beige filmomhulde tabletten met aan één kant "277".
Ondoorzichtige blisterverpakkingen (PVC / PE / PVDC en aluminium).
Verpakkingen van 14, 28, 30, 56, 84, 90 of 98 filmomhulde tabletten en 50 x 1 filmomhulde tabletten in geperforeerde eenheidsdosisblisterverpakkingen.
Mogelijk worden niet alle verpakkingsgrootten in de handel gebracht.
Bron Bijsluiter: AIFA (Italiaans Geneesmiddelenbureau). Inhoud gepubliceerd in januari 2016. De aanwezige informatie is mogelijk niet up-to-date.
Om toegang te hebben tot de meest actuele versie, is het raadzaam om de website van AIFA (Italian Medicines Agency) te bezoeken. Disclaimer en nuttige informatie.
01.0 NAAM VAN HET GENEESMIDDEL
XELEVIA 100 MG TABLETTEN BEDEKT MET FILM
02.0 KWALITATIEVE EN KWANTITATIEVE SAMENSTELLING
Elke tablet bevat sitagliptinefosfaatmonohydraat, overeenkomend met 100 mg sitagliptine.
Voor de volledige lijst van hulpstoffen, zie rubriek 6.1.
03.0 FARMACEUTISCHE VORM
Filmomhulde tablet (tablet).
Ronde, beige filmomhulde tablet met aan één kant "277".
04.0 KLINISCHE INFORMATIE
04.1 Therapeutische indicaties
Voor volwassen patiënten met diabetes mellitus type 2 is Xelevia geïndiceerd om de glykemische controle te verbeteren:
in monotherapie
• bij patiënten die onvoldoende onder controle zijn met alleen dieet en lichaamsbeweging en voor wie metformine niet geschikt is vanwege contra-indicaties of intolerantie.
bij duale orale therapie in combinatie met
• metformine wanneer dieet en lichaamsbeweging plus metformine alleen niet voldoende controle van de bloedglucose geven.
• een sulfonylureumderivaat wanneer dieet en lichaamsbeweging plus de maximaal getolereerde dosis van een sulfonylureumderivaat alleen niet voldoende glykemische controle bieden en wanneer metformine niet geschikt is vanwege contra-indicaties of intolerantie.
• een peroxisoom proliferator-geactiveerde receptor (PPAR?)-agonist (bijv. een thiazolidinedion) wanneer het gebruik van een PPAR-agonist aangewezen is En wanneer dieet en lichaamsbeweging plus PPAR-agonist? alleen zorgen ze niet voor een adequate controle van de bloedglucose.
in drievoudige orale therapie in combinatie met
• een sulfonylureumderivaat en metformine wanneer dieet en lichaamsbeweging plus gecombineerde therapie met deze geneesmiddelen geen adequate glykemische controle bieden.
• een PPAR-agonist? en metformine wanneer het gebruik van een PPAR-agonist geschikt is en wanneer dieet en lichaamsbeweging plus gecombineerde therapie met deze geneesmiddelen geen adequate glykemische controle bieden.
Xelevia is ook geïndiceerd als aanvullende therapie bij insuline (met of zonder metformine) wanneer dieet en lichaamsbeweging plus een stabiele dosis insuline niet voldoende glykemische controle bieden.
04.2 Dosering en wijze van toediening
Dosering
De dosering is eenmaal daags 100 mg sitagliptine. Bij gebruik in combinatie met metformine en/of een PPAR-agonist moet de dosis metformine en/of de PPAR-agonist worden gehandhaafd en moet Xelevia gelijktijdig worden toegediend.
Als Xelevia wordt gebruikt in combinatie met een sulfonylureumderivaat of insuline, kan een lagere dosis van het sulfonylureumderivaat of insuline worden overwogen om het risico op hypoglykemie te verminderen (zie rubriek 4.4).
Als een dosis Xelevia is vergeten, moet deze worden ingenomen zodra de patiënt eraan denkt.
Een dubbele dosis mag niet op dezelfde dag worden ingenomen.
Speciale populaties
Nierschade
Wanneer het gebruik van sitagliptine in combinatie met een ander antidiabetisch geneesmiddel wordt overwogen, moet de wijze van gebruik bij patiënten met een nierfunctiestoornis worden gecontroleerd.
Voor patiënten met een lichte nierfunctiestoornis (creatinineklaring [CrCl] ≥ 50 ml/min) is geen dosisaanpassing vereist.
Voor patiënten met een matige nierfunctiestoornis (CrCl ≥ 30 tot
Voor patiënten met een ernstige nierfunctiestoornis (CrCl-hemodialyse of peritoneale dialyse is de dosis Xelevia eenmaal daags 25 mg. De behandeling kan worden toegediend ongeacht het tijdstip van dialyse.
Aangezien er een dosisaanpassing is op basis van de nierfunctie, wordt aanbevolen de nierfunctie te evalueren voordat de behandeling met Xelevia wordt gestart en periodiek daarna.
leverfunctiestoornis
Er is geen dosisaanpassing nodig voor patiënten met een lichte tot matige leverfunctiestoornis. Xelevia is niet onderzocht bij patiënten met een ernstige leverfunctiestoornis en voorzichtigheid is geboden (zie rubriek 5.2).
Aangezien sitagliptine echter voornamelijk via de nieren wordt geëlimineerd, wordt niet verwacht dat een ernstige leverfunctiestoornis de farmacokinetiek van sitagliptine beïnvloedt.
Bejaarden
Er is geen dosisaanpassing nodig op basis van leeftijd.
Pediatrische populatie
De veiligheid en werkzaamheid van sitagliptine bij kinderen en adolescenten jonger dan 18 jaar. Er zijn geen gegevens beschikbaar.
Wijze van toediening
Xelevia kan met of zonder maaltijden worden ingenomen.
04.3 Contra-indicaties
Overgevoeligheid voor de werkzame stof of voor één van de in rubriek 6.1 vermelde hulpstoffen (zie rubrieken 4.4 en 4.8).
04.4 Bijzondere waarschuwingen en passende voorzorgen bij gebruik
Algemeenheid
Xelevia mag niet worden gebruikt bij patiënten met type I diabetes of voor de behandeling van diabetische ketoacidose.
Acute ontsteking aan de alvleesklier
Het gebruik van dipeptidylpeptidase 4 (DPP-4)-remmers is in verband gebracht met een risico op het ontwikkelen van acute pancreatitis Patiënten moeten worden geïnformeerd over het kenmerkende symptoom van acute pancreatitis: ernstige, aanhoudende buikpijn. Het verdwijnen van pancreatitis is waargenomen na stopzetting van de behandeling. behandeling met sitagliptine (met of zonder ondersteunende behandeling), maar er zijn zeer zeldzame gevallen van necrotiserende of hemorragische pancreatitis en/of overlijden gemeld. Als pancreatitis wordt vermoed, moet de behandeling met Xelevia en andere mogelijk verdachte geneesmiddelen worden stopgezet; als de diagnose acute pancreatitis is bevestigd, mag de behandeling met Xelevia niet opnieuw worden gestart. Voorzichtigheid is geboden bij patiënten met een voorgeschiedenis van pancreatitis.
Hypoglykemie bij gebruik in combinatie met andere antihyperglykemische geneesmiddelen
In klinische onderzoeken met Xelevia als monotherapie en als onderdeel van combinatietherapie met geneesmiddelen waarvan niet bekend is dat ze hypoglykemie veroorzaken (bijv. metformine en/of een PPAR-agonist?), was de incidentie van hypoglykemie gemeld met sitagliptine vergelijkbaar met de incidentie bij patiënten die placebo gebruikten. Hypoglykemie is waargenomen wanneer sitagliptine werd gebruikt in combinatie met insuline of een sulfonylureumderivaat. Daarom kan een lagere dosis sulfonylureumderivaat of insuline worden overwogen om het risico op hypoglykemie te verminderen (zie rubriek 4.2).
Nierschade
Sitagliptine wordt via de nieren uitgescheiden. Om plasmaconcentraties van sitagliptine te bereiken die vergelijkbaar zijn met die bij patiënten met een normale nierfunctie, worden lagere doseringen aanbevolen bij patiënten met matige tot ernstige nierinsufficiëntie, evenals bij patiënten met ESRD die hemodialyse of peritoneale dialyse nodig hebben (zie rubrieken 4.2 en 5.2).
Wanneer het gebruik van sitagliptine in combinatie met een ander antidiabetisch geneesmiddel wordt overwogen, moet de wijze van gebruik bij patiënten met een nierfunctiestoornis worden gecontroleerd.
Overgevoeligheidsreacties
In postmarketingmeldingen zijn ernstige overgevoeligheidsreacties gemeld bij patiënten die met sitagliptine werden behandeld. Deze reacties omvatten anafylaxie, angio-oedeem en exfoliatieve huidaandoeningen, waaronder het Stevens-Johnson-syndroom. Het begin van deze reacties trad op binnen de eerste 3 maanden na het starten van de behandeling, en enkele meldingen traden op na de eerste dosis.
Als een overgevoeligheidsreactie wordt vermoed, moet de behandeling met Xelevia worden stopgezet. Andere mogelijke oorzaken van de gebeurtenis moeten worden onderzocht en er moet een alternatieve behandeling voor diabetes worden gestart.
04.5 Interacties met andere geneesmiddelen en andere vormen van interactie
Effecten van andere geneesmiddelen op sitagliptine
De hieronder beschreven klinische gegevens suggereren dat het risico op klinisch significante interacties met gelijktijdig gebruikte geneesmiddelen beperkt is.
Opleiding in vitro gaf aan dat het belangrijkste enzym dat verantwoordelijk is voor het beperkte metabolisme van sitagliptine CYP3A4 is met een bijdrage van CYP2C8. Bij patiënten met een normale nierfunctie speelt het metabolisme, inclusief dat van CYP3A4, een beperkte rol bij de klaring van sitagliptine. Metabolisme kan een belangrijkere rol spelen bij de eliminatie van sitagliptine in de context van ernstige nierinsufficiëntie of terminale nierziekte (ESRD).Om deze reden is het mogelijk dat krachtige CYP3A4-remmers (bijv. ketoconazol, itraconazol, ritonavir, claritromycine) de de farmacokinetiek van sitagliptine bij patiënten met ernstige nierinsufficiëntie of ESRD De effecten van krachtige CYP3A4-remmers op nierinsufficiëntie zijn niet vastgesteld in een klinisch onderzoek.
Transportstudies in vitro toonde aan dat sitagliptine een substraat is voor p-glycoproteïne e
voor de organische aniontransporter 3 (OAT3). OAT3-gemedieerd transport van sitagliptine werd geremd in vitro probenecide, hoewel het risico op klinisch relevante interacties als beperkt wordt beschouwd. Gelijktijdige toediening van OAT3-remmers is niet geëvalueerd in vivo.
Metformine: Gelijktijdige toediening van meerdere doses metformine 1.000 mg met sitagliptine 50 mg tweemaal daags veranderde de farmacokinetiek van sitagliptine niet significant bij patiënten met type 2-diabetes.
Cyclosporine: Er is een onderzoek uitgevoerd om het effect van ciclosporine, een krachtige remmer van p-glycoproteïne, op de farmacokinetiek van sitagliptine te evalueren. Gelijktijdige toediening van een enkelvoudige orale dosis van 100 mg sitagliptine en een enkelvoudige orale dosis van 600 mg ciclosporine heeft de AUC verhoogd en Cmax van sitagliptine met respectievelijk ongeveer 29% en 68%. Deze veranderingen in de farmacokinetiek van sitagliptine werden niet als klinisch relevant beschouwd. De renale klaring van sitagliptine was niet significant veranderd. Daarom worden geen interacties verwacht. relevant met andere p-glycoproteïneremmers.
Effecten van sitagliptine op andere geneesmiddelen
Digoxine: Sitagliptine had een beperkt effect op de plasmaconcentraties van digoxine. Na toediening van 0,25 mg digoxine gelijktijdig met 100 mg sitagliptine per dag gedurende 10 dagen, nam de plasma-AUC van digoxine met gemiddeld 11% toe en de plasma-Cmax met gemiddeld 18%. Er worden geen dosisaanpassingen van digoxine aanbevolen. De toxiciteit van digoxine moet echter worden gecontroleerd bij patiënten met een risico op digoxinetoxiciteit wanneer sitagliptine en digoxine gelijktijdig worden toegediend.
Gegevens in vitro suggereren dat sitagliptine de CYP450-iso-enzymen niet remt of induceert. In klinische onderzoeken veranderde sitagliptine de farmacokinetiek van metformine, glyburide, simvastatine, rosiglitazon, warfarine of orale anticonceptiva niet significant. in vivo een lage neiging om interacties te veroorzaken met substraten van CYP3A4, CYP2C8, CYP2C9 en met de organische kationtransporter (OCT). Sitagliptine kan een zwakke remmer van p-glycoproteïne zijn in vivo.
04.6 Zwangerschap en borstvoeding
Zwangerschap
Er zijn onvoldoende gegevens over het gebruik van sitagliptine bij zwangere vrouwen Dierstudies hebben reproductietoxiciteit aangetoond bij hoge doses (zie rubriek 5.3) Het potentiële risico voor mensen is niet bekend. Bij gebrek aan gegevens bij de mens mag Xelevia niet tijdens de zwangerschap worden gebruikt.
Voedertijd
Het is niet bekend of sitagliptine wordt uitgescheiden in de moedermelk. Dierstudies hebben de uitscheiding van sitagliptine in de moedermelk aangetoond. Xelevia mag niet worden gebruikt tijdens het geven van borstvoeding.
Vruchtbaarheid
Diergegevens wijzen niet op een effect van de behandeling met sitagliptine op de mannelijke en vrouwelijke vruchtbaarheid. Er is een gebrek aan menselijke gegevens.
04.7 Beïnvloeding van de rijvaardigheid en het vermogen om machines te bedienen
Xelevia heeft geen of een verwaarloosbare invloed op de rijvaardigheid en op het vermogen om machines te bedienen.
Bij het besturen van voertuigen of het bedienen van machines moet er echter rekening mee worden gehouden dat duizeligheid en slaperigheid zijn gemeld.
Bovendien moeten patiënten, wanneer Xelevia wordt gebruikt in combinatie met een sulfonylureumderivaat of met insuline, op het risico van hypoglykemie worden gewezen.
04.8 Bijwerkingen
Samenvatting van het veiligheidsprofiel
Ernstige bijwerkingen, waaronder pancreatitis en overgevoeligheidsreacties, zijn gemeld.
Hypoglykemie is gemeld in verband met sulfonylureumderivaten (4,7% -13,8%) en insuline (9,6%) (zie rubriek 4.4).
Tabel met bijwerkingen
Bijwerkingen worden hieronder vermeld (Tabel 1) per systeem/orgaanklasse en frequentie. Frequenties zijn gedefinieerd als: zeer vaak (≥ 1/10); vaak (≥ 1/100,
Tabel 1. Frequentie van bijwerkingen geïdentificeerd uit placebogecontroleerde klinische onderzoeken met sitagliptine monotherapie en uit postmarketingervaring
* Bijwerkingen die zijn vastgesteld tijdens postmarketingsurveillance.
† Zie rubriek 4.4.
‡ Zie onder TECOS Cardiovasculair veiligheidsonderzoek.
Beschrijving van geselecteerde bijwerkingen
Naast de hierboven beschreven geneesmiddelgerelateerde bijwerkingen waren bijwerkingen die werden gemeld ongeacht het oorzakelijk verband met het geneesmiddel en die optraden in ten minste 5% van de gevallen en het meest bij patiënten die werden behandeld met sitagliptine, waaronder infectie van de bovenste luchtwegen en nasofaryngitis. Bijkomende bijwerkingen gemeld ongeacht oorzakelijk verband met het geneesmiddel die vaker voorkwamen bij patiënten die werden behandeld met sitagliptine (die het 5%-niveau niet bereikten, maar die optraden met een incidentie van> 0,5% hoger met sitagliptine dan die van de controlegroep ) omvatte artrose en pijn in extremiteiten.
Sommige bijwerkingen werden vaker waargenomen in combinatiestudies van sitagliptine met andere antidiabetica dan in studies met monotherapie met sitagliptine, waaronder hypoglykemie (zeer vaak bij de combinatie van sulfonylureumderivaat en metformine), influenza (vaak met insuline (met of zonder metformine)), misselijkheid en braken (vaak met metformine), flatulentie (vaak met metformine of pioglitazon), constipatie (vaak met de combinatie van sulfonylureumderivaat en metformine), perifeer oedeem (vaak met pioglitazon of met de combinatie van pioglitazon en metformine) slaperigheid en diarree (soms met metformine) en droge mond (soms met insuline (met of zonder metformine)).
TECOS Cardiovasculair veiligheidsonderzoek
De studie Trial Evaluating Cardiovascular Outcomes with sitagliptine (TECOS) omvatte 7.332 patiënten die werden behandeld met sitagliptine, 100 mg per dag (of 50 mg per dag als de baseline-eGFR ≥ 30 en 2) en 7.339 patiënten behandeld met placebo in de intentiepopulatie. -traktatie. Beide behandelingen werden toegevoegd aan de therapie die gewoonlijk wordt gebruikt om regionale standaardwaarden voor HbA1c- en CV-risicofactoren te bereiken.De totale incidentie van ernstige bijwerkingen bij met sitagliptine behandelde patiënten was vergelijkbaar met die van met placebo behandelde patiënten
In de intention-to-treat-populatie was de incidentie van ernstige hypoglykemie onder patiënten die insuline en/of een sulfonylureumderivaat gebruikten bij aanvang 2,7% bij patiënten die werden behandeld met sitagliptine en 2,5% bij patiënten die werden behandeld met placebo; bij patiënten die geen insuline gebruikten en/of een basaal sulfonylureumderivaat, was de incidentie van ernstige hypoglykemie 1,0% bij patiënten behandeld met sitagliptine en 0,7% bij patiënten behandeld met placebo. De incidentie van bevestigde diagnoses van pancreatitis was 0,3% bij patiënten die werden behandeld met sitagliptine en 0,2% bij patiënten die werden behandeld met placebo.
Melding van vermoedelijke bijwerkingen
Het melden van vermoedelijke bijwerkingen die optreden na toelating van het geneesmiddel is belangrijk, omdat het een continue controle van de baten/risicoverhouding van het geneesmiddel mogelijk maakt.Beroepsbeoefenaren in de gezondheidszorg wordt verzocht alle vermoedelijke bijwerkingen te melden via het Italiaanse Geneesmiddelenbureau. , website: www.agenziafarmaco.gov.it/it/responsabili.
04.9 Overdosering
Tijdens gecontroleerde klinische onderzoeken bij gezonde proefpersonen werden enkelvoudige doses sitagliptine tot 800 mg toegediend. In één onderzoek werden bij een dosis sitagliptine van 800 mg minimale verhogingen van QTc, die niet als klinisch relevant worden beschouwd, waargenomen. Er is geen ervaring met doses boven 800 mg in klinische onderzoeken. Er werden geen dosisgerelateerde bijwerkingen waargenomen in Fase I-onderzoeken met meerdere doses met doses sitagliptine tot 600 mg per dag gedurende perioden tot 10 dagen en 400 mg per dag gedurende perioden tot 28 dagen.
In het geval van een overdosis is het redelijk om algemene ondersteunende maatregelen te nemen, bijv.: verwijder niet-geabsorbeerd materiaal uit het maagdarmkanaal, gebruik klinische controle (inclusief elektrocardiografie) en stel zo nodig ondersteunende zorg in.
De dialyseerbaarheid van sitagliptine is bescheiden. In klinische onderzoeken werd ongeveer 13,5% van de dosis verwijderd gedurende een hemodialysesessie van 3-4 uur. Langdurige hemodialyse kan worden overwogen indien dit klinisch aangewezen wordt geacht.De dialyseerbaarheid van sitagliptine bij peritoneale dialyse is niet bekend.
05.0 FARMACOLOGISCHE EIGENSCHAPPEN
05.1 Farmacodynamische eigenschappen
Farmacotherapeutische categorie: geneesmiddelen gebruikt bij diabetes, dipeptidylpeptidase 4 (DPP-4) remmers.
ATC-code: A10BH01.
Werkingsmechanisme
Xelevia behoort tot een klasse van orale antihyperglykemische geneesmiddelen die dipeptidylpeptidase 4-remmers (DPP-4) worden genoemd. De verbetering van de glykemische controle die met dit geneesmiddel wordt waargenomen, kan worden gemedieerd door "verhoogde niveaus van actieve incretines. die gedurende de dag uit de darm worden vrijgegeven en hun niveau stijgt als reactie op maaltijden. Incretines maken deel uit van een endogeen systeem dat betrokken is bij de fysiologische regulatie van glucosehomeostase.Wanneer de bloedglucose normaal of verhoogd is, verhogen GLP-1 en GIP de synthese en afgifte van insuline door bètacellen van de pancreas via intracellulaire signaalroutes waarbij cyclisch AMP betrokken is. Het is aangetoond dat behandeling met GLP-1- of DPP-4-remmers in diermodellen van type 2-diabetes de bètacelrespons op glucose verbetert en de biosynthese en afgifte van insuline stimuleert. Bij hogere insulinespiegels wordt de glucoseopname in het weefsel verhoogd GLP-1 vermindert ook de secretie van glucagon door alfacellen van de alvleesklier Lagere glucagonconcentraties, samen met hogere insulinespiegels, resulteren in een verminderde leverproductie van glucose, wat resulteert in een afname van het bloed glucose. De effecten van GLP-1 en GIP zijn glucose-afhankelijk, zodat wanneer de bloedglucose laag is, er geen stimuli voor insulineafgifte en onderdrukking van glucagonsecretie worden waargenomen. Voor zowel GLP-1 als GIP stijgt de stimulatie van insulineafgifte wanneer glucose stijgt Bovendien heeft GLP-1 geen invloed op de normale respons van glucagon op hypoglykemie. De activiteit van GLP-1 en GIP wordt beperkt door het DPP-4-enzym dat incretines snel hydrolyseert tot inactieve metabolieten. Sitagliptine voorkomt de hydrolyse van incretines door DPP-4, waardoor de plasmaconcentraties van de actieve vormen van GLP-1 en GIP toenemen.Verhoging van de actieve incretines sitagliptine verhoogt de insulineafgifte en verlaagt de glucagonspiegels op een glucosemanier.Bij patiënten met type 2 diabetes met hyperglykemie leiden deze veranderingen in insuline- en glucagonspiegels tot een verlaagd hemoglobine A1c (HbA1c) en lagere nuchtere en bloedglucoseconcentraties. post prandium. Het glucoseafhankelijke mechanisme van sitagliptine verschilt van het mechanisme van sulfonylureumderivaten, die de insulinesecretie verhogen, zelfs bij lage glucosespiegels en kunnen leiden tot hypoglykemie bij patiënten met diabetes type 2. Sitagliptine is een krachtige remmer en zeer selectief van DPP-4-enzym en remt niet de activiteit van nauw verwante enzymen DPP-8 of DPP-9 bij therapeutische concentraties.
In een tweedaags onderzoek bij gezonde proefpersonen verhoogde sitagliptine alleen de actieve GLP-1-concentraties, terwijl metformine alleen de actieve en totale GLP-1-concentraties op vergelijkbare wijze verhoogde. Gelijktijdige toediening van sitagliptine en metformine had een additief effect op actieve GLP-1-concentraties. Sitagliptine, maar niet metformine, verhoogde de actieve GIP-concentraties.
Klinische werkzaamheid en veiligheid
Over het algemeen verbeterde sitagliptine de glykemische controle bij toediening als monotherapie of in combinatietherapie (zie tabel 2).
Er zijn twee onderzoeken uitgevoerd om de werkzaamheid en veiligheid van alleen sitagliptine te evalueren. Behandeling met sitagliptine als monotherapie van 100 mg eenmaal daags gaf significante verbeteringen in HbA1c, nuchtere plasmaglucose (FPG) en 2 uur postprandiale glucose (2 uur PPG), vergeleken met placebo in twee onderzoeken, waarvan de ene 18 weken duurde en de andere. 24 weken Verbetering werd waargenomen in surrogaatmarkers van bètacelfunctie, waaronder HOMA-? (Homeostasis Model Assessment-?) hypoglykemie waargenomen bij patiënten die werden behandeld met sitagliptine was vergelijkbaar met placebo. Het lichaamsgewicht nam in de twee onderzoeken niet toe ten opzichte van de uitgangswaarde bij behandeling met sitagliptine, vergeleken met een lichte afname die werd waargenomen bij met placebo behandelde patiënten.
Sitagliptine 100 mg eenmaal daags induceerde significante verbeteringen in glykemische parameters in vergelijking met placebo in twee 24 weken durende add-on studies met sitagliptine, één in combinatie met metformine en één in combinatie met pioglitazon. De verandering in lichaamsgewicht ten opzichte van baseline was vergelijkbaar voor patiënten die werden behandeld met sitagliptine in vergelijking met placebo. In deze onderzoeken werd "een" vergelijkbare incidentie van hypoglykemie gemeld bij patiënten die werden behandeld met sitagliptine of placebo.
Een 24 weken durende placebogecontroleerde studie werd opgezet om de werkzaamheid en veiligheid te evalueren van sitagliptine (100 mg eenmaal daags) toegevoegd aan glimepiride alleen of aan glimepiride in combinatie met metformine.De toevoeging van sitagliptine of glimepiride alleen of met glimepiride en metformine veroorzaakte significante verbeteringen in glycemische parameters. Patiënten die met sitagliptine werden behandeld, hadden een bescheiden gewichtstoename in vergelijking met degenen die placebo kregen.
Een 26 weken durende, placebogecontroleerde studie werd opgezet om de werkzaamheid en veiligheid te evalueren van sitagliptine (100 mg eenmaal daags) toegevoegd aan de combinatie van pioglitazon en metformine. De toevoeging van sitagliptine aan pioglitazon en metformine resulteerde in significante verbeteringen in de glykemische parameters. De verandering in lichaamsgewicht ten opzichte van baseline was vergelijkbaar bij patiënten die werden behandeld met sitagliptine en bij degenen die werden behandeld met placebo. De incidentie van hypoglykemie was ook vergelijkbaar bij de patiënten behandeld met placebo patiënten behandeld met sitagliptine of placebo.
Een 24 weken durende placebogecontroleerde studie werd opgezet om de werkzaamheid en veiligheid te evalueren van sitagliptine (100 mg eenmaal daags) toegevoegd aan insuline (in een stabiele dosis gedurende ten minste 10 weken) met of zonder metformine (ten minste 1.500 mg). Bij patiënten die voorgemengde insuline gebruikten, was de gemiddelde dagelijkse dosis 70,9 E/dag Bij patiënten die niet-voorgemengde (middellangwerkende/langwerkende) insuline gebruikten, was de gemiddelde dagelijkse dosis 44,3 E/dag. De toevoeging van sitagliptine aan insuline veroorzaakte significante verbeteringen in de glykemische parameters. Er was in geen van beide groepen een significante verandering in lichaamsgewicht ten opzichte van baseline.
In een 24 weken durende, placebogecontroleerde, factoriële studie met combinatietherapie bij aanvang, resulteerde sitagliptine 50 mg tweemaal daags in combinatie met metformine (500 mg of 1.000 mg tweemaal daags) in significante verbeteringen in de glykemische parameters in vergelijking met elke monotherapie. met de combinatie sitagliptine en metformine was vergelijkbaar met wat werd waargenomen met alleen metformine of met placebo; er werd geen verandering ten opzichte van de uitgangswaarde waargenomen bij patiënten die werden behandeld met sitagliptine als monotherapie. De incidentie van hypoglykemie was vergelijkbaar tussen de behandelingsgroepen.
Tabel 2: HbA1c-resultaten in onderzoeken met placebogecontroleerde monotherapie en combinatietherapie *
* Alle behandelde patiëntenpopulatie (intention-to-treatment-analyse).
† Kleinste-kwadratengemiddelden aangepast voor eerdere hypoglykemische therapie en uitgangswaarde.
‡ P
§ HbA1c (%) na 18 weken. HbA1c (%) na 24 weken.
# HbA1c (%) na 26 weken.
¶ Kleinste kwadratengemiddelden gecorrigeerd voor metforminegebruik bij bezoek 1 (ja / nee), voor insulinegebruik bij bezoek 1
[voorgemengd versus niet-voorgemengd (middellangwerkend of langwerkend)] en basale waarde.Behandelingsinteracties per laag (voor metformine- en insulinegebruik) waren niet significant (p> 0,10).
Een 24 weken durende actief-gecontroleerde studie (metformine) werd opgezet om de werkzaamheid en veiligheid van sitagliptine 100 mg eenmaal daags (N = 528) te evalueren in vergelijking met metformine (N = 522) bij patiënten die geen adequate glykemische controle hadden met dieet en inspanning en die geen antihyperglykemische therapie kregen (minstens 4 maanden zonder therapie). De gemiddelde dosis metformine was ongeveer 1.900 mg per dag. De verlaging van HbA1c ten opzichte van de gemiddelde uitgangswaarden van 7,2% was -0,43% voor sitagliptine en -0,57% voor metformine (analyse per protocol) De totale incidentie van gastro-intestinale bijwerkingen die als geneesmiddelgerelateerd werden beschouwd bij patiënten die werden behandeld met sitagliptine was 2, 7% vergeleken met 12,6% bij metformine behandelde patiënten.
De incidentie van hypoglykemie was niet significant verschillend tussen de behandelingsgroepen (sitagliptine, 1,3%; metformine, 1,9%). Het lichaamsgewicht nam in beide groepen af ten opzichte van de uitgangswaarde (sitagliptine, -0,6 kg; metformine -1,9 kg).
In een onderzoek waarin de werkzaamheid en veiligheid werden vergeleken van toevoeging van sitagliptine 100 mg eenmaal daags of glipizide (een sulfonylureumderivaat) bij patiënten met onvoldoende glykemische controle op metformine als monotherapie, was sitagliptine vergelijkbaar met glipizide bij het verlagen van HbA1c. De gemiddelde dosis glipizide die in de vergelijkingsgroep werd gebruikt, was 10 mg/dag, waarbij ongeveer 40% van de patiënten gedurende het onderzoek een dosis glipizide van ≤ 5 mg/dag nodig had. Patiënten in de sitagliptinegroep ondervonden echter meer stopzettingen vanwege een gebrek aan werkzaamheid dan in de glipizidegroep. Patiënten die met sitagliptine werden behandeld, vertoonden een significante gemiddelde afname van het lichaamsgewicht ten opzichte van de uitgangswaarde vergeleken met een significante gewichtstoename die werd gezien bij patiënten die glipizide kregen (-1,5 vs +1,1kg). In deze studie verbeterde de pro-insuline/insuline-ratio, een marker van insulinesynthese en afgifte-efficiëntie, met sitagliptine en verslechterd met behandeling met glipizide.De incidentie van hypoglykemie in de sitagliptinegroep (4,9%) was significant lager dan die in de glipizidegroep (32,0%).
Een 24 weken durende placebogecontroleerde studie met 660 patiënten werd opgezet om de insulinesparende werkzaamheid en veiligheid te evalueren van sitagliptine (100 mg eenmaal daags) toegevoegd aan insuline glargine met of zonder metformine (minstens 1.500 mg) tijdens intensivering van de insulinetherapie. Baseline HbA1c was 8,74% en baseline insulinedosis was 37 IE/dag. Patiënten kregen de instructie om de dosis insuline glargine te titreren op basis van nuchtere glucosewaarden gemeten met een vingerprik. In week 24 was de verhoging van de dagelijkse insulinedosis 19 IE/dag bij patiënten behandeld met sitagliptine en 24 IE/dag bij patiënten behandeld met placebo.De verlaging van HbA1c bij patiënten behandeld met sitagliptine en insuline (met of zonder metformine) was - 1,31% versus -0,87% bij patiënten behandeld met placebo en insuline (met of zonder metformine), een verschil van -0,45% [95% BI: -0,60, -0,29].De incidentie van hypoglykemie was 25,2% bij patiënten behandeld met sitagliptine en insuline (met of zonder metformine) en 36,8% bij patiënten die werden behandeld met placebo en insuline (met of zonder metformine). Het verschil was voornamelijk te wijten aan een hoger percentage patiënten in de placebogroep die 3 of meer episoden van hypoglykemie doormaakten (9,4 versus 19,1%). Er was geen verschil in de incidentie van ernstige hypoglykemie.
Bij patiënten met een matige tot ernstige nierfunctiestoornis werd een onderzoek uitgevoerd waarin sitagliptine 25 of 50 mg eenmaal daags werd vergeleken met glipizide 2,5 tot 20 mg/dag. Bij deze studie waren 423 patiënten met chronische nierinsufficiëntie betrokken (geschatte glomerulaire filtratiesnelheid
Een ander onderzoek waarin sitagliptine 25 mg eenmaal daags en glipizide 2,5 tot 20 mg/dag werd vergeleken, werd uitgevoerd bij 129 patiënten met ESRD die dialyse ondergingen. Na 54 weken was de gemiddelde afname van HbA1c vanaf baseline -0,72% met sitagliptine en -0,87% met glipizide. In deze studie was het werkzaamheids- en veiligheidsprofiel van sitagliptine 25 mg eenmaal daags over het algemeen vergelijkbaar met dat waargenomen in andere monotherapiestudies die werden uitgevoerd bij patiënten met een normale nierfunctie. De incidentie van hypoglykemie was niet significant verschillend tussen de behandelingsgroepen (sitagliptine, 6,3%; glipizide, 10,8%).
In een ander onderzoek onder 91 patiënten met diabetes type 2 en chronische nierinsufficiëntie (creatinineklaring)
TECOS was een gerandomiseerde studie bij 14.671 patiënten in de intention-to-treat-populatie met HbA1c-waarden variërend van ≥6,5 tot 8,0% en met vastgestelde CV-ziekte die werden behandeld met sitagliptine (7.332) 100 mg per dag (of 50 mg per dag als baseline eGFR was ≥30 en 2) of placebo (7.339) toegevoegd aan therapie die gewoonlijk wordt gebruikt om regionale standaardwaarden voor HbA1c- en CV-risicofactoren te bereiken Patiënten met eGFR 2 werden naar verwachting niet in het onderzoek opgenomen De onderzoekspopulatie omvatte 2.004 patiënten ≥75 jaar en 3.324 patiënten met nierinsufficiëntie (eGFR 2).
In de loop van het onderzoek was het totale geschatte gemiddelde (SD) verschil in HbA1c tussen de sitagliptine- en placebogroep 0,29%, 95% BI (-0,32, -0,27); P
Het primaire cardiovasculaire eindpunt was een samenstelling van vroeg optredende cardiovasculaire sterfte, niet-fataal myocardinfarct, niet-fatale beroerte of ziekenhuisopname wegens instabiele angina. Secundaire cardiovasculaire eindpunten waren onder meer vroege aanvang van cardiovasculaire sterfte, niet-fataal myocardinfarct of niet- fatale beroerte, aanvang van de afzonderlijke componenten van het samengestelde primaire eindpunt; dood door welke oorzaak dan ook; en ziekenhuisopnames voor congestief hartfalen.
Na een mediane follow-up van drie jaar verhoogde sitagliptine, indien toegevoegd aan de gewoonlijk gebruikte therapie, het risico op ernstige cardiovasculaire bijwerkingen of het risico op ziekenhuisopname voor hartfalen niet in vergelijking met therapie die gewoonlijk zonder sitagliptine wordt gebruikt bij patiënten met type diabetes. 2 (Tabel 3).
Tabel 3. Percentages samengestelde cardiovasculaire uitkomsten en belangrijkste uitkomsten
Ondergeschikt
* De incidentie per 100 patiëntjaren wordt berekend als 100 × (totaal aantal patiënten met ≥ 1 voorval tijdens de in aanmerking komende blootstellingsperiode voor de totale patiëntjaren van follow-up).
† Gebaseerd op een regionaal gestratificeerd Cox-model. Voor samengestelde eindpunten komt de p-waarde overeen met een non-inferioriteitstest om aan te tonen dat de hazard ratio kleiner is dan 1,3. Voor alle andere eindpunten komt de p-waarde overeen met een test voor verschillen in risicoverhoudingen.
‡ Analyse van ziekenhuisopname voor hartfalen werd bij aanvang aangepast voor anamnestische voorgeschiedenis van hartfalen.
Pediatrische populatie
Het Europees Geneesmiddelenbureau heeft besloten tot uitstel van de verplichting voor de fabrikant om de resultaten in te dienen van onderzoek met Xelevia in een of meerdere subgroepen van pediatrische patiënten met type 2 diabetes mellitus (zie rubriek 4.2 voor informatie over pediatrisch gebruik).
05.2 Farmacokinetische eigenschappen
Absorptie
Na orale toediening van een dosis van 100 mg aan gezonde proefpersonen werd sitagliptine snel geabsorbeerd, met piekplasmaconcentraties (mediane Tmax) 1 tot 4 uur na de dosis, de gemiddelde plasma-AUC van sitagliptine was 8, 52 M • uur, Cmax was 950 nM De absolute biologische beschikbaarheid van sitagliptine is ongeveer 87% Aangezien gelijktijdige toediening van een vetrijke maaltijd met sitagliptine geen effect had op de farmacokinetiek, kan Xelevia onafhankelijk van maaltijden worden toegediend.
De plasma-AUC van sitagliptine nam dosisproportioneel toe.Dosisproportionaliteit werd niet vastgesteld voor Cmax en C24h (Cmax nam meer toe dan dosisproportionaliteit en C24h nam in mindere mate toe met betrekking tot dosisproportionaliteit).
Verdeling
Het gemiddelde distributievolume bij steady-state na een enkelvoudige intraveneuze dosis sitagliptine van 100 mg aan gezonde proefpersonen is ongeveer 198 liter. De fractie sitagliptine die op reversibele wijze aan plasma-eiwitten wordt gebonden is laag (38%).
Biotransformatie
Sitagliptine wordt voornamelijk onveranderd via de urine uitgescheiden en het metabolisme is een ondergeschikte metabolische route. Ongeveer 79% van sitagliptine wordt onveranderd in de urine uitgescheiden.
Na een orale dosis [14C] sitagliptine werd ongeveer 16% van de radioactiviteit uitgescheiden als metabolieten van sitagliptine. Er zijn sporen van zes metabolieten van sitagliptine gevonden en deze zullen naar verwachting niet bijdragen aan de plasma-DPP-4-remmende activiteit van sitagliptine. in vitro gaf aan dat het enzym dat primair verantwoordelijk is voor het beperkte metabolisme van sitagliptine CYP3A4 is, met een bijdrage van CYP2C8.
Gegevens in vitro toonde aan dat sitagliptine geen remmer is van CYP-isozymen CYP3A4, 2C8, 2C9, 2D6, 1A2, 2C19 of 2B6 en geen inductor is van CYP3A4 en CYP1A2.
Eliminatie
Na orale toediening van [14C] sitagliptine aan gezonde proefpersonen werd ongeveer 100% van de toegediende radioactiviteit binnen één week na toediening uitgescheiden in de feces (13%) of urine (87%). De terminale aPPAR t½ na een orale dosis sitagliptine van 100 mg was ongeveer 12,4 uur. Sitagliptine accumuleert slechts minimaal bij meerdere doses. De nierklaring was ongeveer 350 ml/min.
Eliminatie van sitagliptine vindt voornamelijk plaats via renale excretie en omvat actieve tubulaire secretie. Sitagliptine is een substraat voor de humane organische aniontransporter 3 (hOAT-3) die mogelijk betrokken is bij de renale eliminatie van sitagliptine. De klinische relevantie van hOAT-3 bij het transport van sitagliptine is niet vastgesteld. Sitagliptine is ook een substraat van p-glycoproteïne, dat ook betrokken kan zijn bij het mediëren van de renale eliminatie van sitagliptine. Ciclosporine, een p-glycoproteïneremmer, verminderde echter niet de renale klaring van sitagliptine. Sitagliptine is geen substraat voor OCT2- of OAT1- of PEPT½-transporters. In vitro, remde sitagliptine het door OAT3 (IC50 = 160 M) of p-glycoproteïne (tot 250 M) gemedieerde transport bij therapeutisch relevante plasmaconcentraties niet.In een klinisch onderzoek had sitagliptine een beperkt effect op de plasmaconcentraties van digoxine, wat erop wijst dat sitagliptine een zwakke remmer van p-glycoproteïne kan zijn.
Kenmerken van patiënten
De farmacokinetiek van sitagliptine was over het algemeen vergelijkbaar bij gezonde proefpersonen en bij patiënten met type 2-diabetes.
Nierschade
Er werd een open-label onderzoek met een enkelvoudige dosis uitgevoerd om de farmacokinetiek van een verlaagde dosis sitagliptine (50 mg) te evalueren bij patiënten met een verschillende mate van chronische nierinsufficiëntie in vergelijking met normale gezonde controlepersonen. De studie omvatte patiënten met een nierfunctiestoornis die volgens de creatinineklaring als licht (50 tot
Patiënten met een lichte nierfunctiestoornis hadden geen klinisch significante verhogingen van de plasmaconcentraties van sitagliptine in vergelijking met normale gezonde controlepersonen. Een ongeveer 2-voudige toename van de plasma-AUC van sitagliptine werd waargenomen bij patiënten met een matige nierfunctiestoornis, en bij patiënten met ernstige nierfunctiestoornis en ESDR die hemodialyse ondergaan, werd een ongeveer 4-voudige toename van de plasma-AUC waargenomen in vergelijking met gezonde controlepersonen. Sitagliptine werd in beperkte mate verwijderd door hemodialyse (13,5% over een hemodialysesessie van 3 tot 4 uur die 4 uur na de dosis begon). patiënten met matige en ernstige nierinsufficiëntie, evenals bij patiënten met ESRD die dialyse nodig hebben (zie rubriek 4.2).
leverfunctiestoornis
Er is geen dosisaanpassing nodig voor Xelevia bij patiënten met een lichte of matige leverfunctiestoornis (Child-Pugh-score ≤ 9). Er is geen klinische ervaring bij patiënten met een ernstige leverfunctiestoornis (Child-Pugh-score > 9) Aangezien sitagliptine voornamelijk via de nieren wordt geëlimineerd, wordt echter niet verwacht dat een ernstige leverfunctiestoornis de farmacokinetiek van sitagliptine beïnvloedt.
Bejaarden
Er is geen dosisaanpassing nodig op basis van leeftijd Leeftijd had geen klinisch significant effect op de farmacokinetiek van sitagliptine op basis van gegevens van een fase I en fase II farmacokinetische populatieanalyse Bij ouderen (van 65 tot 80 jaar), ongeveer 19% hoger plasma concentraties sitagliptine werden waargenomen dan bij jonge mensen.
Pediatrische populatie
Er zijn geen onderzoeken uitgevoerd met Xelevia bij pediatrische patiënten.
Andere kenmerken van patiënten
Er is geen dosisaanpassing nodig op basis van geslacht, ras of body mass index (BMI). Deze kenmerken hadden geen klinisch significant effect op de farmacokinetiek van sitagliptine op basis van gegevens van een Fase I samengestelde farmacokinetische analyse en gegevens van een Fase I en Fase II farmacokinetische populatieanalyse.
05.3 Gegevens uit het preklinisch veiligheidsonderzoek
Bij knaagdieren werd nier- en levertoxiciteit waargenomen bij systemische blootstellingswaarden gelijk aan 58 keer de menselijke blootstelling, terwijl het niveau zonder effect werd gevonden bij 19 keer de menselijke blootstelling. Snijtandafwijkingen werden waargenomen bij ratten bij blootstellingsniveaus gelijk aan 67 maal de klinische blootstelling bij de mens; het niveau zonder effect voor deze gebeurtenis was 58-voudig op basis van een 14 weken durende rattenstudie. De relevantie van deze gegevens voor mensen is niet bekend Voorbijgaande lichamelijke symptomen die verband houden met de behandeling zijn waargenomen bij honden bij blootstellingsniveaus van ongeveer 23 keer het klinische blootstellingsniveau, waarvan sommige wijzen op neurale toxiciteit, zoals ademen met open mond, speekselvloed, wit schuimig braken, ataxie, tremor, verminderde activiteit en/of gebogen houding. Bij doses gelijk aan ongeveer 23 maal het systemische blootstellingsniveau bij de mens, werd histologisch ook zeer milde tot milde skeletspierdegeneratie waargenomen.Een niveau zonder effect voor deze gebeurtenissen werd gevonden bij een blootstelling gelijk aan 6 maal het klinische blootstellingsniveau.
Sitagliptine toonde geen genotoxiciteit aan in preklinische onderzoeken. Sitagliptine was niet kankerverwekkend bij muizen. Bij ratten was er een verhoogde incidentie van leveradenomen en -carcinomen bij systemische blootstellingsniveaus gelijk aan 58 maal de blootstelling bij de mens. Deze verhoogde incidentie van levertumoren bij de rat is waarschijnlijk secundair aan de chronische levertoxiciteit die optreedt bij deze hoge doses.
Vanwege de grote veiligheidsmarge (19 keer op dit niveau zonder effect), worden deze neoplastische laesies niet relevant geacht voor de blootstellingsomstandigheden bij mensen.
Er werden geen nadelige effecten op de vruchtbaarheid waargenomen bij mannelijke en vrouwelijke ratten die voor en tijdens de paring met sitagliptine werden behandeld.
In pre-/postnatale ontwikkelingsstudies bij ratten lieten sitagliptine geen bijwerkingen zien.
Reproductietoxiciteitsstudies lieten een lichte, behandelingsgerelateerde toename zien van de incidentie van foetale ribmisvormingen (afwezige, hypoplastische en golvende ribben) bij de nakomelingen van ratten bij systemische blootstellingsniveaus die 29 keer hoger zijn dan die bij de mens. Maternale toxiciteit werd waargenomen bij konijnen bij blootstellingsniveaus die hoger waren dan 29 maal de blootstellingsniveaus bij de mens.Vanwege de brede veiligheidsmarges wijzen deze bevindingen niet op de aanwezigheid van relevante reproductieve risico's bij de mens. Sitagliptine wordt in aanzienlijke hoeveelheden uitgescheiden in de melk van zogende ratten (melk/plasmaverhouding: 4: 1).
06.0 FARMACEUTISCHE INFORMATIE
06.1 Hulpstoffen
Tabletkern:
microkristallijne cellulose (E460),
watervrij calciumwaterstoffosfaat (E341),
croscarmellosenatrium (E468),
magnesiumstearaat (E470b),
natriumstearylfumaraat
Tabletcoating:
poly (vinylalcohol),
macrogol 3350,
talk (E553b),
titaandioxide (E171),
rood ijzeroxide (E172),
geel ijzeroxide (E172)
06.2 Incompatibiliteit
Niet relevant.
06.3 Geldigheidsduur
3 jaar
06.4 Speciale voorzorgsmaatregelen bij bewaren
Voor dit geneesmiddel zijn er geen speciale bewaarcondities.
06.5 Aard van de primaire verpakking en inhoud van de verpakking
Ondoorzichtige blisterverpakkingen (PVC / PE / PVDC en aluminium). Verpakkingen van 14, 28, 30, 56, 84, 90 of 98 filmomhulde tabletten en 50 x 1 filmomhulde tabletten in geperforeerde eenheidsdosisblisterverpakkingen.
Mogelijk worden niet alle verpakkingsgrootten in de handel gebracht.
06.6 Instructies voor gebruik en verwerking
Ongebruikte medicijnen en afval afkomstig van dit medicijn moeten worden weggegooid in overeenstemming met de lokale regelgeving.
07.0 HOUDER VAN DE VERGUNNING VOOR HET IN DE HANDEL BRENGEN
Merck Sharp & Dohme Ltd.
Hertford Road, Hoddesdon
Hertfordshire EN11 9BU
VK
08.0 NUMMER VAN DE VERGUNNING VOOR HET IN DE HANDEL BRENGEN
EU / 1/07/382/013
037794132
EU / 1/07/382/014
037794144
EU / 1/07/382/015
037794157
EU / 1/07/382/016
037794169
EU / 1/07/382/017
037794171
EU / 1/07/382/018
037794183
EU / 1/07/382/023
037794233
EU / 1/07/382/024
037794245
09.0 DATUM VAN EERSTE VERGUNNING OF VERLENGING VAN DE VERGUNNING
Datum eerste vergunning: 21 maart 2007
Datum van de meest recente verlenging: 20 januari 2012
10.0 DATUM VAN HERZIENING VAN DE TEKST
28 januari 2016

















---livello-avanzato.jpg)