Algemeenheid
Osteogenesis imperfecta is een aangeboren genetische ziekte, niet gekoppeld aan geslacht, die verantwoordelijk is voor een zekere botfragiliteit en een duidelijke neiging tot fracturen.
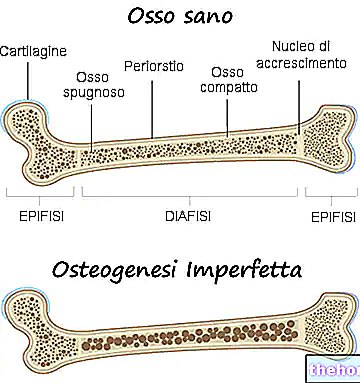
De symptomen van osteogenesis imperfecta zijn talrijk; over het algemeen bestaan ze uit: botverzwakking, sterke neiging tot botbreuken, aanwezigheid van blauwe, grijze of paarse oculaire sclerae, aanwezigheid van botmisvormingen of andere skeletveranderingen, driehoekig gezicht, tandfragiliteit, enz. .
Over het algemeen zijn de volgende zaken essentieel voor een juiste diagnose van osteogenesis imperfecta: lichamelijk onderzoek, medische geschiedenis, medische beeldvormingstests, een type I collageenbeoordelingstest en een genetische test.
Helaas zijn momenteel de enige behandelingen die beschikbaar zijn voor patiënten met osteogenesis imperfecta symptomatisch. De ziekte in kwestie is in feite ongeneeslijk.
Wat is osteogenesis imperfecta?
Osteogenesis imperfecta is een genetische ziekte die de botten van de aangedane persoon zwakker maakt en vatbaarder maakt voor breuken.
In werkelijkheid verwijzen artsen met de term osteogenesis imperfecta naar een heterogene groep genetische ziekten, gekenmerkt door een zekere mate van botfragiliteit. Er zijn daarom verschillende vormen (of typen) van osteogenesis imperfecta, sommige veel ernstiger dan andere.
HET IS EEN AANgeboren ZIEKTE
Bij mensen die er last van hebben, is osteogenesis imperfecta een ziekte die vanaf de geboorte aanwezig is en daarom kan het in alle opzichten worden gedefinieerd als een aangeboren ziekte.
IS HET SEKSGERELATEERD?
Osteogenesis imperfecta is geen geslachtsgebonden genetische ziekte, zoals hemofilie of het syndroom van Klinefelter.
EPIDEMILOGIE
Volgens sommige statistische onderzoeken zou de incidentie van osteogenesis imperfecta gelijk zijn aan één geval per 15.000-20.000 geboorten. Dit betekent dat elke 15.000-20.000 pasgeborenen er een hebben die is aangetast door osteogenesis imperfecta.
Andere statistische studies hebben ook aangetoond dat osteogenesis imperfecta zowel mannen als vrouwen treft, en dat het geen voorkeur heeft voor een bepaalde populatie of etnische groep.
Levensduur is een uiterst variabele parameter, die afhangt van de vorm van osteogenesis imperfecta.
Oorzaken
Osteogenesis imperfecta is bijna altijd het gevolg van een kwalitatieve en kwantitatieve wijziging van de productie van type I collageen.
Type I collageen is essentieel voor het versterken van botten en voor het behoud van gezonde bindweefsels die kraakbeen, pezen, huid, oculaire sclera, enz. vormen.
Een verandering in de productie van collageen type I beïnvloedt daarom de sterkte van de botten en de goede gezondheid van de bindweefsels die in het menselijk lichaam aanwezig zijn.
WAT VERANDERT DE COLLAGEENPRODUCTIE?
Een genetische ziekte is een aandoening die ontstaat door een mutatie van een of meer genen die het cellulaire DNA vormen.
Bij osteogenesis imperfecta zijn de oorzaken hiervan vrijwel altijd te vinden in de mutatie van één of beide genen COL1A1 (gelegen op chromosoom 17) en COL1A2 (gelegen op chromosoom 7).
Onder normale omstandigheden reguleren COL1A1 en COL1A2 de normale productie van type I collageen; in aanwezigheid van mutaties onder hun hoede, falen ze in hun regulerende functie.
Belangrijk: welke andere genen, indien gemuteerd, veroorzaken osteogenesis imperfecta?
Naast de mutaties van COL1A1 en COL1A2 zijn mutaties in de genen IFITM5, SERPINF1, CRTAP en LEPRE1 mogelijke oorzaken van osteogenesis imperfecta.
De bovengenoemde genen bestrijken functies die anders zijn dan COL1A1 en COL1A2 - daarom controleren ze niet de productie van type I collageen - maar ze hebben nog steeds een "invloed op de sterkte en weerstand van de botten van het menselijk skelet.
WAT VOOR EEN GENETISCHE ZIEKTE IS HET?
Osteogenesis imperfecta is een autosomaal erfelijke ziekte.
De term autosomaal, geassocieerd met een genetische ziekte, geeft aan dat de aandoening in kwestie te wijten is aan genetische mutaties op basis van autosomale en niet-geslachtschromosomen.
De lezers worden eraan herinnerd dat de mens een chromosomale set van 23 paren totale chromosomen bezit, waarvan 22 paren van het autosomale type en slechts één paar van het seksuele type. Het paar chromosomen van het seksuele type beïnvloedt het geslacht van de individueel.
Osteogenesis imperfecta volgend op mutaties in COL1A1, COL1A2 en IFITM5 heeft alle kenmerken van een autosomaal dominante ziekte.Als het te wijten is aan mutaties in de genen SERPINF1, CRTAP en LEPRE1, heeft het de kenmerken van een autosomaal recessieve ziekte.
TYPES
Momenteel geloven artsen dat er 8 soorten (of vormen) osteogenesis imperfecta zijn. Om de verschillende typen te onderscheiden, besloten ze de Romeinse nummering te gebruiken, om precies te zijn de eerste acht Romeinse cijfers.
Onderstaande tabel toont de 8 vormen van osteogenesis imperfecta, de mutaties die ze veroorzaken en andere kenmerken.
Vent
gemuteerd gen
Type genetische ziekte
DE
COL1A1
Autosomaal dominant
II
COL1A1 en COL1A2
Autosomaal dominant
III
COL1A1 en COL1A2
Autosomaal dominant
NS
COL1A1 en COL1A2
Autosomaal dominant
V.
IFITM5
Autosomaal dominant
JIJ
SERPINF1
Autosomaal recessief
VII
CRTAP
Autosomaal recessief
VIII
HAAS 1
Autosomaal recessief
* NB: de mutaties in COL1A1 en COL1A2, die de eerste vier vormen van osteogenesis imperfecta veroorzaken, zijn uiteraard genetische veranderingen met iets andere kenmerken. Anders zou het geen zin hebben om de een van de ander te onderscheiden.
Symptomen, tekenen en complicaties
Alle soorten osteogenesis imperfecta zijn verantwoordelijk voor een verzwakking van de botten, zodat de persoon die door de ziekte wordt getroffen, een bijzondere aanleg heeft voor fracturen. De mate van verzwakking van de botten is afhankelijk van de vorm; voor sommige hiervan is deze verzwakking groter dan voor andere.
Dit gezegd hebbende, moet erop worden gewezen dat elke vorm van osteogenesis imperfecta zijn eigen symptomatische beeld heeft, dat voor sommigen zich het symptomatologische beeld van andere vormen kan herinneren.
MOGELIJKE SYMPTOMEN EN TEKENS
De mogelijke symptomen en tekenen van osteogenesis imperfecta zijn onder meer:
- Aanwezigheid van botmisvormingen;
- Aanwezigheid van een kort en klein lichaam (bedoeld als romp);
- Gewrichtsproblemen (bijv. losse gewrichten);
- Spier zwakte;
- Blauwe, paarse of grijze oculaire sclera;
- Driehoekig gezicht;
- Vat borst;
- Morfologische afwijkingen van de wervelkolom;
- tandheelkundige kwetsbaarheid;
- Achteruitgang of totaal gehoorverlies;
- Ademhalingsproblemen
- Problemen in verband met de afwezigheid of afwezigheid van type 1 collageen.
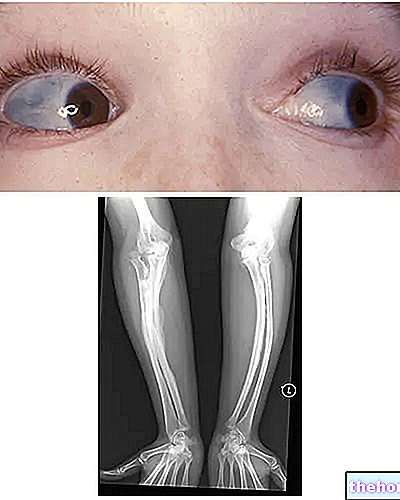
Osteogenesis Imperfecta: let op de blauwe verkleuring van de sclerae en de botdeformaties die de ziekte kenmerken. Van wikipedia.org
WAT ZIJN DE MEEST ERNSTIGE VORMEN VAN IMPERFECTE OSTEOGENESE?
Artsen classificeren de symptomatologische ernst van de verschillende soorten osteogenesis imperfecta op een schaal van 3 graden, namelijk: de milde graad, de matige graad en de ernstige graad.
Slechts één vorm behoort tot de categorie "milde graad": "type I osteogenesis imperfecta"; 4 vormen van osteogenesis imperfecta behoren tot de categorie "matige graad": IV, V en VI; tot slot behoren tot de categorie "ernstige graad" 3 vormen: II, III, VII en VIII.
TYPE I: KENMERKEN
De meest voorkomende en minst ernstige vorm van allemaal, type I osteogenesis imperfecta, heeft de volgende kenmerken:
- Het veroorzaakt fracturen, vooral vóór de puberteit;
- Het heeft "bijna geen invloed op de lengte, dus patiënten hebben meestal een normale" lengte;
- Veroorzaakt gewrichtsproblemen en spierzwakte
- Het is verantwoordelijk voor blauwe, paarse of grijze sclera;
- Het is de oorzaak van driehoekige gezichts- en spinale anomalieën;
- Het veroorzaakt bijna nooit botmisvormingen. Als het ze provoceert, zijn ze minimaal;
- Het kan tandfragiliteit en/of gehoorverlies veroorzaken (dit laatste treedt meestal op op volwassen leeftijd);
- Het wordt geassocieerd met de aanwezigheid van type I collageen, dat normaal van kwaliteit is maar abnormaal in kwantiteit (het is slechter dan normaal).
TYPE II: KENMERKEN
Type II osteogenesis imperfecta wordt gekenmerkt door:
- Doodsoorzaak bij de geboorte of kort daarna. Ademhalingsproblemen veroorzaken bijna altijd de dood;
- Aanwezigheid van aanzienlijke botfragiliteit en ernstige botmisvormingen;
- Korte gestalte en onderontwikkelde longen
- Blauw, paars of grijs gekleurde sclera;
- Aanwezigheid van kwantitatieve en kwalitatieve afwijkingen van type I collageen.
TYPE III: KENMERKEN
Type III osteogenesis imperfecta heeft de volgende kenmerken:
- Hoewel het zeer ernstig is, veroorzaakt het niet vaak de dood in de neonatale periode;
- Het wordt geassocieerd met "hoge botfragiliteit;
- Het is verantwoordelijk voor kleine gestalte, gewrichtsproblemen, spierzwakte (vooral in de benen en armen), borstkas, driehoekig gezicht en abnormale kromming van de wervelkolom;
- Het is te wijten aan blauwe, paarse of grijze sclera;
- Het kan ademhalingsproblemen, broosheid van de tanden en gehoorverlies veroorzaken;
- Het is vaak verantwoordelijk voor botmisvormingen;
- Het wordt geassocieerd met kwalitatieve en kwantitatieve afwijkingen van type I collageen.
TYPE IV: KENMERKEN
Type IV osteogenese wordt gekenmerkt door:
- Een mate van botfragiliteit tussen vormen II en III en vorm I;
- Korter dan gemiddeld postuur;
- Blauw, paars of grijs gekleurde sclera;
- Botvervormingen van milde / matige entiteit, lichte afwijkingen van de wervelkolom en de borstkas;
- Driehoekig gezicht;
- Mogelijke aanwezigheid van tandfragiliteit en gehoorverlies;
- Aanwezigheid van type I collageenafwijkingen.
TYPE V: KENMERKEN
Type V osteogenesis imperfecta lijkt in sommige opzichten op type IV osteogenesis imperfecta. Het heeft echter enkele eigenaardigheden, namelijk:
- Normaal gekleurde sclera;
- Afwezigheid van tandheelkundige kwetsbaarheid;
- Vorming van abnormale benige eelt, tijdens het genezingsproces van gebroken botten;
- Verkalking van het interossale membraan dat zich tussen de radius en de ulna bevindt. Dit belemmert de mobiliteit van de onderarm.
TYPE VI: KENMERKEN
Ook osteogenesis imperfecta van type VI is vergelijkbaar met vorm IV. Om het van de laatste te onderscheiden, zijn enkele eigenaardigheden, waaronder de hoge bloedspiegels van alkalische fosfatase en de aanwezigheid, op sommige botten, van lamellen (benige) vergelijkbaar met stekels van vissen.
TYPE VII: KENMERKEN
Symptomatisch kan type VII osteogenesis imperfecta in sommige omstandigheden op type IV lijken en in andere omstandigheden op type II.
De eigenaardigheden van deze ernstige pathologische vorm zijn onder meer:
- Korte gestalte;
- De aanwezigheid van een extreem korte humerus (armbeen) en dijbeen (dijbeen);
- De frequente aanwezigheid van een heupafwijking, bekend als coxa vara.
TYPE VIII: KENMERKEN
Type VIII osteogenesis imperfecta doet sterk denken aan vormen II en III.
Onder zijn eigenaardige kenmerken vallen de volgende op: de ernstige groeiachterstand, de ernstige hypomineralisatie van het skelet en de afwezigheid (of schaarse aanwezigheid) van het prolyl-3-hydroxylase-enzym.
Diagnose
Over het algemeen begint het diagnostisch proces dat patiënten met een vermoedelijke vorm van osteogenesis imperfecta ondergaan met een zorgvuldig lichamelijk onderzoek en een zorgvuldige medische voorgeschiedenis; dan gaat het verder met een "analyse van de familiegeschiedenis van de patiënt en met een reeks diagnostische beeldvormingstesten (röntgenfoto's, CT-scans, enz.); ten slotte eindigt het met een kwantitatieve en kwalitatieve evaluatie van type I collageen en met een genetische test.
Tegenwoordig bestaat de mogelijkheid om osteogenesis imperfecta te diagnosticeren, zelfs in de prenatale fase, door een zwangere vrouw aan een echografie te onderwerpen.
HET BELANG VAN HET OBJECTIEVE ONDERZOEK EN DE GESCHIEDENIS
Een arts die deskundig is op het gebied van osteogenesis imperfecta is vaak in staat om de bovengenoemde ziekte te diagnosticeren, zelfs alleen door middel van lichamelijk onderzoek en anamnese. Dit betekent dat deze diagnostische tests van niet te verwaarlozen belang zijn.
EVALUATIE VAN TYPE I COLLAGEENPRODUCTIE
In de regel is de kwalitatieve en kwantitatieve evaluatie van type I collageen een zeer betrouwbare test, aangezien, zoals gezegd, de meeste gevallen van osteogenesis imperfecta worden gekenmerkt door mutaties in de genen die de productie van type 1 collageen regelen.
Om de kwantiteit en kwaliteit van type I collageen dat aanwezig is op cellulair niveau in een persoon te beoordelen, kunnen artsen vertrouwen op een huidbiopsie of een bepaald bloedonderzoek.
Beide evaluatieve tests zijn behoorlijk complex en de patiënt (of zijn ouders) moet mogelijk enkele weken wachten om de resultaten te kennen.
GENETISCHE TEST
Door een genetische test die het volledige DNA van het onderzochte individu onderzoekt, kunnen artsen de kenmerken van de aanwezige genetische mutatie definitief vaststellen.
Over het algemeen is de uitvoering van een genetische test op al het cellulaire DNA voorzien wanneer de evaluatie van de kenmerken van type I collageen niet de gewenste resultaten heeft opgeleverd, of wanneer het geen mutatie in COL1A1 of COL1A2 is die de "osteogenesis imperfecta" veroorzaakt.
PRENATALE DIAGNOSE
Prenatale echografie is zeer nuttig bij het identificeren van type II en type III osteogenesis imperfecta.
Therapie
Er is momenteel geen specifieke remedie voor osteogenesis imperfecta, met andere woorden, mensen met osteogenesis imperfecta zijn voorbestemd om met de bovengenoemde aandoening te leven tot de dood, die vaak te wijten is aan de gevolgen van de ziekte zelf.
Het ontbreken van specifieke therapie sluit het bestaan van andere vormen van behandeling niet uit. In feite zijn onder de therapeutische mogelijkheden van een patiënt met osteogenesis imperfecta verschillende symptomatische therapieën inbegrepen; met symptomatische therapieën bedoelen we behandelingen die de symptomen kunnen verlichten, het ziekteverloop kunnen vertragen en de ernstigste gevolgen kunnen voorkomen (of op zijn minst uitstellen).
MOGELIJKE SYMPTOMATISCHE BEHANDELINGEN
In de lijst met mogelijke symptomatische behandelingen voor osteogenesis imperfecta vallen de volgende op:
- Het chirurgisch inbrengen, in de langste botten (NB: het meest vatbaar voor breuken), van nagels die een grotere weerstand bieden tegen breuken en misvormingen. Deze operatie heet rodding intramedullair;
- Conservatieve of chirurgische behandeling van fracturen en/of botafwijkingen;
- Tandheelkundige zorg, om de gezondheid van het gebit te waarborgen;
- Pijnstillende therapieën bij zeer pijnlijke meervoudige fracturen;
- Fysiotherapie, voor spierverlenging en -versterking Een elastisch en tonisch spierapparaat stelt u in staat vallen te voorkomen die tot verschillende botbreuken kunnen leiden;
- Het gebruik van hulpmiddelen voor het voortbewegen, waaronder rolstoelen, beugels, krukken, enz.
VOORDELEN VAN BEWEGING
Voor personen met osteogenesis imperfecta raden artsen aan om constant aan lichaamsbeweging en beweging in het algemeen te doen, aangezien beide activiteiten bijdragen aan de versterking van het skelet- en spierstelsel.
Tot de aanbevolen sporten behoren: zwemmen, aangezien het een "lichamelijke activiteit met weinig impact op het skelet" is, en wandelen.
VOORDELEN VAN EEN GEZONDE LEVENSSTIJL
Een gezond leven leiden, roken vermijden, te veel alcohol drinken, te veel en slecht eten, enz., heeft meer dan discrete gezondheidsvoordelen voor patiënten met osteogenesis imperfecta, aangezien het de progressie van de ziekte vertraagt en de botfragiliteit vermindert.
SYMPTOMATISCHE BEHANDELINGEN IN DE EXPERIMENTATIEFASE
Momenteel evalueren artsen en onderzoekers de effectiviteit van sommige symptomatische behandelingen, waaronder groeihormoonbehandeling en op bisfosfonaten gebaseerde intraveneuze en orale therapie.
Op dit moment voorspellen de resultaten van de bovengenoemde onderzoeksbehandelingen veel goeds voor de hele medische gemeenschap.
Prognose
Osteogenesis imperfecta is een ziekte met een negatieve prognose, aangezien deze ongeneeslijk is, de kwaliteit van leven drastisch in gevaar brengt en in sommige gevallen voortijdige dood van de getroffen persoon veroorzaakt.
Er moet echter worden opgemerkt dat, mede dankzij moderne symptomatische behandelingen, veel mensen met een milde vorm van osteogenesis imperfecta een aangenaam en bevredigend leven kunnen leiden.
preventie
Helaas is er momenteel geen preventieve maatregel tegen osteogenesis imperfecta.

















.jpg)


