Algemeenheid
Cystische fibrose is de meest voorkomende autosomaal recessieve ziekte bij de blanke bevolking en treft ongeveer 1 op de 2500 personen.
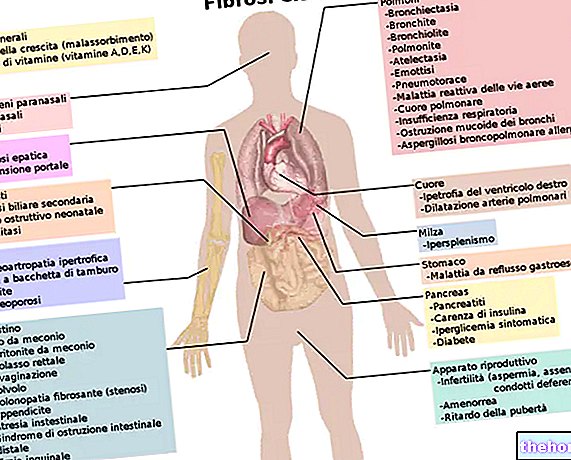
Deze pathologische aandoening staat bekend om zijn schadelijke effecten op het ademhalingssysteem, maar het tast ook andere systemen aan, zoals het spijsverterings- en voortplantingssysteem.
Bij personen met cystische fibrose zijn de luchtwegen verstopt met dik en stroperig slijm, dat zelfs met de meest krachtige hoest moeilijk te verwijderen is. Ademen wordt moeilijk en patiënten - als er niet voortdurend inspanningen worden gedaan om de luchtwegen meerdere keren per dag schoon te houden - lopen het risico te overlijden aan hun eigen afscheiding. Patiënten met taaislijmziekte sterven vaak aan longontsteking, omdat verstopte luchtwegen een vruchtbare omgeving bieden voor bacteriën om te groeien.

Oorzaken
Cystic fibrosis wordt veroorzaakt door mutaties in het cystic fibrosis transmembrane conductance regulator (CFTR) gen op chromosoom 7 (locus mapping: 7q31).
Er zijn minstens 1.500 mutaties van het CFTR-gen bekend. De meest voorkomende mutatie wordt gewoonlijk "Delta-F508" (DF508) genoemd en wordt veroorzaakt door de deletie van 3 basenparen in exon 10, wat resulteert in het verlies van fenylalanine op positie 508.
Het eiwit dat wordt gecodeerd door het CFTR-gen is een transmembraankanaal dat behoort tot de superfamilie van verkeers-ATPasen of ABC-transporters, gelegen op het niveau van het apicale membraan van epitheelcellen en verantwoordelijk voor het transport van het chloorion.
Onder normale omstandigheden scheiden bepaalde cellen die de luchtwegen bekleden slijm af samen met een waterige vloeistof die de dichtheid ervan vermindert. Bij cystische fibrose is de afscheiding van de waterige vloeistof sterk verminderd, waardoor het slijm erg dik wordt en moeilijk uit de luchtwegen te verwijderen is.
In het respiratoire epitheel, zoals alle vloeistofdragende epitheel, hangt het transport van water af van het transport van opgeloste stoffen. Om water af te scheiden, transporteren de cellen van het respiratoire epitheel actief chloorionen (Cl-) van de interstitiële vloeistof naar het lumen, waardoor een negatief elektrisch potentiaal ontstaat dat een passieve stroom van natrium (Na +) in dezelfde richting veroorzaakt. Na+ en Cl - ze verhogen de osmotische druk van de vloeistof die de zijde van het epitheel die naar het lumen is gericht, bevochtigt, als gevolg daarvan beweegt het water passief volgens de osmotische gradiënt, van de interstitiële vloeistof naar het lumen. cystische fibrose verhindert het transport van Cl- direct en indirect interfereert met het transport van Na+ en water, waardoor de osmotische gradiënt die nodig is voor de afscheiding van water in het epitheel niet wordt gecreëerd.
Risicofactoren
- Familie erfenis. Aangezien cystische fibrose een erfelijke ziekte is, die op een autosomaal recessieve manier wordt overgedragen, is het belangrijk om rekening te houden met de familiegeschiedenis (anamnese) van de toekomstige ouders.
Dus als kinderen slechts één kopie erven (slechts één zieke ouder), zullen ze geen cystische fibrose ontwikkelen, maar zullen ze asymptomatische dragers zijn en mogelijk het defecte gen doorgeven aan hun kinderen. Zoals te zien is in de afbeelding, is er een kans van één op vier (25%) dat het kind wordt getroffen door cystische fibrose (heterozygoot voor het CFTR-gen en dus slechts één kopie van abnormale genen dragen), zoals blijkt uit de afbeelding. homozygoot voor het CFTR-gen).
- Bevolking van erbij horen. De incidentie van cystische fibrose is hoger bij mensen van Noord- en Europese afkomst.

Klinische symptomen en tekenen
Voor meer informatie: Cystic Fibrosis Symptomen
De ernst van de symptomen kan variëren, afhankelijk van het verloop van de ziekte: de meeste klinische symptomen hebben betrekking op de luchtwegen en het maag-darmstelsel.