Wat is het Marfan-syndroom?
Het Marfan-syndroom beschrijft een complexe erfelijke aandoening van het bindweefsel, die voornamelijk de ogen, het cardiovasculaire systeem en het bewegingsapparaat aantast. Aangezien elk orgaan echter uit bindweefsel bestaat, kan het Marfan-syndroom idealiter de groei en functie van elke anatomische plaats vernietigen en ernstig verstoren.
Het syndroom wordt overgedragen als een autosomaal dominante eigenschap: we hebben dus te maken met een ernstige genetische ziekte, met een extreem variabele "fenotypische expressie (de defecten kunnen enorm verschillen van familie tot familie of van patiënt tot patiënt).
Wat het Marfan-syndroom veroorzaakt, is de wijziging van het FBN1-gen (op chromosoom 15), dat codeert voor fibrilline-1, een zeer belangrijk bindglycoproteïne dat de structurele ondersteuning vormt voor microfibrillen.
Microfibrillen: bestaande uit fibrilline, microfibrillen zijn aanwezig in de extracellulaire matrix, waarin ze een verstrengeling vormen voor de afzetting van elastine in de elastische vezels. Hoewel alomtegenwoordig in het lichaam, zijn microfibrillen vooral in overvloed aanwezig in de aorta, ligamenten en zonules van de ciliaire lichamen (op oculair niveau).
Aangezien dit een autosomaal dominante ziekte is, worden alleen kinderen die een veranderd FBN-1-gen van beide ouders hebben geërfd, getroffen door het Marfan-syndroom. Toch is de ziekte in een op de vier gevallen het gevolg van spontane mutaties bij patiënten zonder familiegeschiedenis.
De naam van de ziekte is afgeleid van de Franse kinderarts die het voor het eerst beschreef in 1896 (A. Marfan), waarna het nodig was om tot 1991 te wachten om het veranderde gen te identificeren dat betrokken was bij de symptomatologische manifestatie: de ontdekker was F. Ramirez.
Bekijk de video
- Bekijk de video op youtube
Oorzaken
We hebben vermeld dat het syndroom van Marfan de onmiddellijke uitdrukking is van de mutatie van een gen dat codeert voor fibrilline-1.
FIBRILLIN 1 is een glycoproteïnecomponent van elastine, essentieel voor het verzekeren en behouden van de elasticiteit en sterkte van het weefsel. Onder fysiologische omstandigheden bindt fibrilline 1 aan een ander eiwit, bekend als TGF-bèta (of transformerende groeifactor beta). TGF-bèta lijkt betrokken te zijn bij schadelijke processen die de vasculaire gladde spieren en de extracellulaire matrix beïnvloeden. Uitgaande van deze veronderstellingen zijn sommige auteurs ervan overtuigd dat het Marfan-syndroom, naast de mutatie van het FBN-1-gen, ook te wijten is aan een overmaat aan TGF-bèta, vooral in de aorta, hartkleppen en longen.

incidentie
Naar schatting treft het Marfan-syndroom 1 op de 3.000-5.000 geboorten en komt het zonder onderscheid voor tussen mannen en vrouwen. Statistieken tonen aan dat 75% van de patiënten een positieve familiegeschiedenis heeft; bij de overige 25% ligt de oorzaak in sporadische mutaties die op de een of andere manier verband lijken te houden met de hoge leeftijd van de vader op het moment van conceptie.
Kinderen met extreem ernstige vormen van het Marfan-syndroom hebben een 'levensverwachting van minder dan een jaar'.
Vóór de ontwikkeling van openhartchirurgische strategieën hadden de meeste patiënten met het Marfan-syndroom een gemiddelde levensverwachting van 32 jaar; dankzij de constante verbetering van medische en farmacologische therapieën leven momenteel patiënten met het Marfan-syndroom gemiddeld tot 60 jaar.
Tekenen en symptomen
Voor meer informatie: Symptomen van het Marfan-syndroom
Het Marfan-syndroom kan volledig asymptomatisch optreden. Getroffen patiënten hebben een overdreven slanke structuur en zijn onevenredig lang en dun. De onderste en bovenste ledematen zijn veel langer dan de romp (dolicostenomegalie). Er is ook sprake van arachnodactylie om het concept van de overdreven lengte van de vingers, typisch voor degenen die lijden aan het Marfan-syndroom, het beste uit te drukken: de handen worden daarom vergeleken met de benen van een spin.
Qua lengte hebben deze patiënten een gestalte met een gemiddelde boven het 97e percentiel.
Naast de andere onderscheidende kenmerken die vaak aanwezig zijn bij patiënten met het Marfan-syndroom, herinneren we ons ook:
- Opening van de armen groter dan de hoogte
- Losse gewrichten → overdreven gewrichtsmobiliteit
- Misvorming van de borstwand
- Verplaatsing van de lens
- Bovenlichaam minder ontwikkeld dan het onderlichaam
- Spontane pneumothorax (11%)
- Scoliose
- Huidstrepen ter hoogte van de dij, rug, deltaspier, borstvinnen
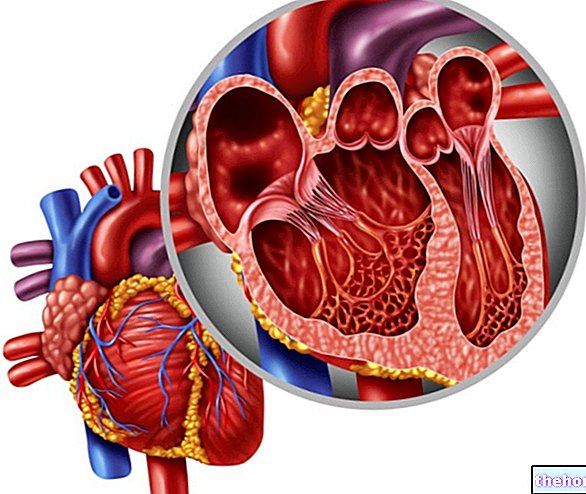
Een van de meest problematische symptomen die verband houden met het Marfan-syndroom, herinneren we ons de verzakking van de hartklep en de insufficiëntie van de mitralisklep: een vergelijkbare aandoening kan gemakkelijk de verwijding van de aortaring en aortadissectie bevorderen.
De tabel geeft de symptomen weer die bij patiënten met het Marfan syndroom te vinden zijn. De daar beschreven personages zijn niet altijd aanwezig, maar een groot deel ervan is terug te vinden.
Mogelijke symptomen
Huid
Striae in het thoracale, lumbale en sacrale gebied
Ogen
Visusverandering, astigmatisme, netvliesloslating, geslotenhoekglaucoom, lensluxatie, bijziendheid
Botstructuur
Artralgie, kyfoscoliose, dolichostenomelie (overmatige ontwikkeling in lengte van de ledematen ten opzichte van de romp), hypermobiliteit, hoog gehemelte, misvormde borstkas, platvoeten, strakke en dunne polsen, abnormale terugkeer/uitsteeksel van het borstbeen, scoliose, gebogen schouders, spondylolisthesis
vingers
arachnodactylie
longen
Spontane pneumothorax, dyspneu, idiopathische obstructieve longziekte
Gezichtsveranderingen
Ogivaal gehemelte (misvorming van het gehemelte), mandibulaire retrognathie (ontwikkelingsdefect van de kaak), langwerpig gezicht
Hart
Angina pectoris, abdominaal aorta-aneurysma, hartritmestoornissen, thoracale aorta dilatatie/ruptuur/dissectie, aorta-insufficiëntie, mitralisklepprolaps
Taal
Moeite met spraak
Diagnose
Gezien de meer dan 200 mogelijke mutaties is het gebruik van genetische markers voor diagnostische doeleinden bijna onmogelijk.
De beoordeling van het syndroom van Marfan is niet altijd zo direct, omdat de fenotypische expressie van de mutatie niet altijd duidelijk en gemakkelijk te identificeren is. De diagnostische vertraging kan de overleving van de patiënt ernstig in gevaar brengen: denk maar aan het niet herkennen van een cardiovasculair probleem.
De diagnostische criteria voor het Marfan syndroom zijn in 1996 internationaal opgesteld: de diagnose bestaat uit het "onderzoek naar de familiegeschiedenis geassocieerd met een combinatie van grote en kleine indicatoren van het syndroom.
Enkele van de vele diagnostische tests die worden gebruikt, zijn:
- echocardiogram
- magnetische angioresonantie en CT (voor het onderzoek van de aorta)
- magnetische resonantie angiografie (MRA) met contrastvloeistof (om de interne structuren van de aorta te markeren)
- onderzoek met spleetlampen (om de mogelijke dislocatie van de lens te analyseren)
- oogdrukmeting (om de mogelijke aanwezigheid van glaucoom te benadrukken)
- genetische tests (aanbevolen voordat een kind wordt verwekt om vast te stellen of het syndroom al dan niet aanwezig is)
therapieën
Aangezien dit een genetische ziekte is, is er geen medicijn of behandeling die de ziekte kan omkeren.
Het gebruik van medicijnen is echter essentieel om de symptomen te verlichten en eventuele complicaties, met name hartcomplicaties, te voorkomen.Hiervoor zijn bloeddrukverlagende medicijnen, zoals sartanen (vooral), ACE-remmers en bètablokkers bijzonder geschikt.
In het kader van het Marfan-syndroom kunnen patiënten die ook lijden aan scoliose een specifieke behandeling volgen, evenals voor degenen die getroffen zijn door glaucoom.
Chirurgie is denkbaar om de abnormale aortadilatatie te corrigeren, een element dat vaak de meerderheid van de patiënten met het Marfan-syndroom verenigt.
Verder: Marfan Syndroom - Medicijnen en behandeling "