Algemeenheid
Retinoblastoom (Rb) is een kwaadaardige oogtumor die ontstaat uit de cellen van het netvlies. Deze kanker kan op elke leeftijd voorkomen, maar het begin komt het meest voor tijdens de kindertijd vóór de leeftijd van vijf.

Kanker bij kinderen is agressief: retinoblastoom kan zich verspreiden naar lymfeklieren, botten of beenmerg. Zelden betreft het het centrale zenuwstelsel (hersenen en ruggenmerg).
Ongeveer 90% van de kinderen met retinoblastoom heeft een positieve prognose (waarschijnlijkheid op genezing), op voorwaarde dat de diagnose vroeg wordt gesteld en de behandeling wordt gestart voordat de kanker zich uitbreidt. Waar mogelijk is het doel van medische interventie om het gezichtsvermogen van de patiënt te behouden.
Oorzaken
De reeks gebeurtenissen die leidt tot het ontstaan van tumoren is complex en begint wanneer cellen in het netvlies een mutatie (of deletie) ontwikkelen waarbij het RB1-tumorsuppressorgen betrokken is, dat zich op de q14-band van chromosoom 13 (13q14) bevindt.
Elke cel heeft normaal gesproken twee RB1-genen:
- Als ten minste één kopie van het gen correct werkt, ontstaat er geen retinoblastoom (maar neemt het risico toe);
- Wanneer beide kopieën van het gen zijn gemuteerd of ontbreken, treedt ongecontroleerde celproliferatie op.
In veel gevallen is het onduidelijk wat precies veranderingen in het RB1-gen veroorzaakt (sporadisch retinoblastoom); deze kunnen het gevolg zijn van willekeurige genetische fouten, die bijvoorbeeld optreden tijdens reproductie en celdeling. Het is echter bekend dat de genetische afwijkingen die ten grondslag liggen aan retinoblastoom ook kunnen worden doorgegeven van ouders op kinderen, met een autosomaal dominant overervingspatroon. Dit betekent dat als een ouder een gemuteerd (dominant) gen draagt, elk kind 50% kans heeft om het te erven en 50% kans op een normale genetische samenstelling (recessieve genen).
- Een enkele cel inactiveert zijn enige normale kopie van het RB1-gen (één kopie is al gemuteerd);
- Het verlies van de twee exemplaren van RB1 leidt tot een "buitensporige proliferatie van het netvlies.
- Een enkele cel inactiveert een van zijn normale RB1-genen;
- De tweede kopie van het RB1-gen is geïnactiveerd;
- Het verlies van de twee kopieën van RB1 veroorzaakt een overmatige celproliferatie die leidt tot retinoblastoom.
Genetische en moleculaire kenmerken
- Retinoblastoom was de eerste tumor die direct werd geassocieerd met een "genetische afwijking (deletie of mutatie van de q14-band van chromosoom 13).
- RB1 codeert voor het pRb-eiwit, dat een sleutelrol speelt in de celcyclus: het maakt DNA-replicatie en celcyclusprogressie mogelijk, omdat het deelneemt aan de transcriptiecontrole van S-fase-genen (G1 → † "S).
- Naast retinoblastoom is het RB1-gen geïnactiveerd bij blaas-, borst- en longkanker.
Erfelijk retinoblastoom
Kinderen met erfelijk retinoblastoom hebben de neiging om de ziekte op jongere leeftijd te ontwikkelen dan sporadische gevallen. Bovendien lopen deze kinderen een verhoogd risico op andere niet-oculaire kankers, aangezien de afwijking in het RB1-gen aangeboren is (dwz aanwezig vanaf de geboorte) en alle cellen in het lichaam aantast (bekend als een kiembaanmutatie), inclusief die van beide. netvliezen: om deze reden hebben kinderen met de erfelijke vorm vaak bilateraal retinoblastoom in plaats van slechts één oog.
Symptomen
Voor meer informatie: Symptomen van retinoblastoom
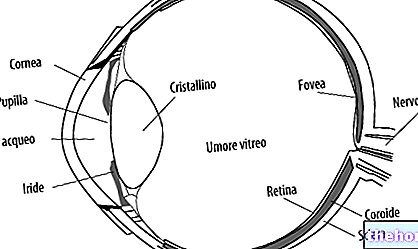
Het meest voorkomende en voor de hand liggende teken van retinoblastoom is het abnormale uiterlijk van de pupil, die een grijswitte reflectie vertoont wanneer deze wordt geraakt door een lichtstraal (leukocoria of amaurotische kattenreflex). Andere tekenen en symptomen zijn: verminderd gezichtsvermogen, oogpijn en roodheid en ontwikkelingsachterstand. Sommige kinderen met retinoblastoom kunnen scheelzien (niet goed uitgelijnde ogen); in andere gevallen is het mogelijk om neovasculair glaucoom te vinden, dat na enige tijd vergroting van het oog (buftalmo) kan veroorzaken.
Kankercellen kunnen het oog en andere structuren verder binnendringen:
- Intraoculair retinoblastoom. Retinoblastoom kan worden gedefinieerd als intraoculair wanneer de tumor zich volledig in het oog bevindt. Het neoplasma kan alleen in het netvlies worden gevonden of ook andere delen aantasten, zoals het vaatvlies, het corpus ciliare en een deel van de oogzenuw. Intraoculair retinoblastoom wordt daarom niet verspreid naar de weefsels rond de buitenkant van het oog.
- Extraoculair retinoblastoom.De tumor kan zich vermenigvuldigen en de weefsels rond het oog aantasten (orbitaal retinoblastoom). De kanker kan zich ook verspreiden naar andere delen van het lichaam, zoals de hersenen, de wervelkolom, het beenmerg en de lymfeklieren (gemetastaseerd retinoblastoom).
De aanwezigheid van orbitale extensie, oogbetrokkenheid en invasie van de oogzenuw zijn bekende risicofactoren voor de ontwikkeling van gemetastaseerd retinoblastoom.
Diagnose
Bij een positieve familieanamnese ondergaat de patiënt regelmatig oogonderzoeken voor screening op kanker. Als congenitaal retinoblastoom bilateraal is, wordt het meestal gediagnosticeerd in het eerste levensjaar, terwijl wanneer het slechts één oog aantast, de aanwezigheid van de tumor kan worden bevestigd op een leeftijd van ongeveer 18-30 maanden.
De klinische diagnose van retinoblastoom wordt gesteld door onderzoek van de fundus.De tumor kan, afhankelijk van de locatie, zichtbaar worden tijdens een eenvoudig onderzoek van het oog, door middel van indirecte oftalmoscopie. Beeldvormingstechnieken kunnen worden gebruikt om de diagnose te bevestigen, de stadiëring van de tumor te bepalen (waar deze is, hoe wijdverbreid deze is, of deze de functies van andere organen in het lichaam aantast, enz.) en om te bepalen of de behandeling effectief is geweest . Onderzoeken kunnen echografie, computertomografie (CT) en magnetische resonantiebeeldvorming (MRI) omvatten.
De moleculair-genetische diagnose is mogelijk door de identificatie van de mutatie van het RB1-gen. De cytogenetische analyse (dwz van de chromosomen) van perifere bloedlymfocyten wordt gebruikt om deleties of herschikkingen van chromosoom 13 (13q14.1-q14.2) op te sporen. .
behandelingen
In het geval van retinoblastoom kunnen verschillende behandelingsopties worden gebruikt.
De doelen van de behandeling zijn:
- Elimineer de tumor en red het leven van de patiënt;
- Bewaar het oog indien mogelijk;
- behoud het zicht zoveel mogelijk;
- Vermijd de ontwikkeling van andere vormen van kanker, die ook door behandeling kunnen worden veroorzaakt, vooral bij kinderen met erfelijk retinoblastoom.
Prognose (kans op herstel) en behandelingsopties zijn afhankelijk van de volgende factoren:
- Stadium van de tumor;
- leeftijd van de patiënt en algemene gezondheidstoestand;
- Locatie, grootte en aantal tumorfoci;
- Verspreiding van de kanker naar andere gebieden dan de oogbol
- Hoe waarschijnlijk is het dat het gezichtsvermogen in één of beide ogen behouden kan blijven.
De meeste gevallen van retinoblastoom worden vroeg gediagnosticeerd en met succes behandeld, voordat de kanker buiten de oogbol kan uitzaaien, wat resulteert in een genezingspercentage van meer dan 90%.



-a-cosa-serve.jpg)