Actieve ingrediënten: Erytropoëtine (epoëtine alfa)
EPREX 2.000 IE/ml, 4.000 IE/ml, 10.000 IE/ml en 40.000 IE/ml OPLOSSING VOOR INJECTIE in VOORGEVULDE SPUITEN
Waarom wordt Eprex gebruikt? Waar is het voor?
Wat is EPREX en waar is het voor?
EPREX bevat de werkzame stof epoëtine alfa, een eiwit dat het beenmerg stimuleert om meer rode bloedcellen aan te maken, cellen die hemoglobine (een stof die zuurstof kan transporteren) vervoeren.Epoëtine alfa is een kopie van humaan erytropoëtine en werkt als zodanig.
- EPREX wordt gebruikt voor de behandeling van symptomatische anemie veroorzaakt door nierfalen.
- bij kinderen die hemodialyse ondergaan
- bij volwassenen die hemodialyse en peritoneale dialyse ondergaan
- bij volwassenen met ernstige anemie die nog geen dialyse ondergaan.
Als u nierfalen heeft en uw nieren niet genoeg erytropoëtine produceren (noodzakelijk voor de aanmaak van rode bloedcellen), heeft u mogelijk weinig rode bloedcellen in uw bloed. EPREX wordt voorgeschreven om het beenmerg te stimuleren om meer rode bloedcellen aan te maken.
- EPREX wordt gebruikt voor de behandeling van bloedarmoede die kan ontstaan tijdens chemotherapie voor solide tumoren, maligne lymfoom of multipel myeloom (beenmergkanker), als uw arts denkt dat u mogelijk een bloedtransfusie nodig heeft.EPREX kan de behoefte aan transfusies verminderen.
- EPREX wordt gebruikt voor de behandeling van patiënten met matige anemie die in aanmerking komen om hun bloed te deponeren in afwachting van een operatie, zodat het tijdens of na de operatie kan worden getransfundeerd.Omdat EPREX de aanmaak van rode bloedcellen stimuleert, is het mogelijk om bij deze mensen bloed af te nemen. een grotere hoeveelheid bloed.
- EPREX wordt gebruikt voor de behandeling van matig anemische volwassen patiënten die een grote orthopedische operatie ondergaan (bijv. heup- of knievervanging) om de noodzaak van bloedtransfusies te verminderen.
Contra-indicaties Wanneer Eprex niet mag worden gebruikt
Gebruik geen EPREX
- Als u allergisch (overgevoelig) bent voor epoëtine alfa of voor één van de andere bestanddelen van EPREX (vermeld onder Inhoud van de verpakking en overige informatie);
- Als bij u de diagnose "Pure Red Cell Aplasia" (het beenmerg kan niet genoeg rode bloedcellen maken) is vastgesteld na een eerdere behandeling met een stof die de aanmaak van rode bloedcellen stimuleert (inclusief EPREX). Zie rubriek Mogelijke bijwerkingen.
- Als u problemen heeft met ongecontroleerde hoge bloeddruk
- Om de aanmaak van rode bloedcellen te stimuleren (zodat er meer bloed bij u kan worden afgenomen) als u tijdens of na de operatie geen transfusies van uw eigen bloed kunt krijgen.
- Als u een electieve grote orthopedische operatie moet ondergaan (bijv. heup- of knievervanging) en:
- heeft een ernstige hartziekte
- u ernstige problemen heeft met uw aderen en slagaders
- onlangs een hartaanval of beroerte heeft gehad
- kan geen bloedverdunnende medicijnen nemen
EPREX is mogelijk niet geschikt voor u, bespreek dit dan met uw arts. Tijdens het gebruik van EPREX kunnen sommige mensen geneesmiddelen nodig hebben om het risico op bloedstolsels te verminderen.Als u geen anticoagulantia kunt gebruiken, mag u EPREX niet gebruiken.
Voorzorgen bij gebruik Wat u moet weten voordat u Eprex inneemt
Wees extra voorzichtig met EPREX
EPREX en andere rode bloedcelstimulerende middelen kunnen bij alle patiënten het risico op het ontwikkelen van bloedstolsels verhogen. Dit risico kan hoger zijn als u andere risicofactoren heeft voor het ontwikkelen van bloedstolsels (bijvoorbeeld als u in het verleden een bloedstolsel heeft gehad of overgewicht heeft, diabetes heeft, een hartaandoening heeft of gedurende lange tijd geïmmobiliseerd bent geweest vanwege operatie of ziekte). Vertel uw arts over een van deze dingen. Uw arts zal u helpen beslissen of EPREX geschikt voor u is.
Het is belangrijk dat u met uw arts praat als een van de volgende situaties op u van toepassing is.
U kunt EPREX nog steeds gebruiken, maar bespreek dit eerst met uw arts.
- Als u weet dat u pijn heeft of geleden heeft aan:
- Hoge bloeddruk;
- Toevallen of toevallen
- Leverziekte;
- Bloedarmoede door andere oorzaken;
- Porfyrie (een zeldzame bloedziekte).
- Als u kanker heeft, moet u zich ervan bewust zijn dat stoffen die de aanmaak van rode bloedcellen stimuleren (zoals EPREX) als een groeifactor kunnen werken en in theorie de tumorprogressie kunnen beïnvloeden. Afhankelijk van uw individuele toestand kan een bloedtransfusie de voorkeur hebben. Bespreek dit met uw arts.
Let vooral op stoffen die de aanmaak van rode bloedcellen stimuleren:
EPREX behoort tot een groep stoffen die de aanmaak van rode bloedcellen stimuleren, zoals menselijke erytropoëtische factoren. De arts zal er altijd voor zorgen dat de exacte naam van het product dat hij gebruikt, wordt genoteerd. Als u tijdens uw behandeling een stof krijgt die tot dezelfde groep behoort maar verschilt van EPREX, neem dan voor gebruik contact op met uw arts of apotheker.
Interacties Welke medicijnen of voedingsmiddelen kunnen het effect van Eprex . veranderen?
EPREX heeft normaal gesproken geen invloed op andere geneesmiddelen, maar vertel het uw arts altijd als u andere geneesmiddelen gebruikt of kort geleden heeft gebruikt, inclusief geneesmiddelen waarvoor geen recept nodig is.
Als u een geneesmiddel gebruikt dat ciclosporine wordt genoemd (dat bijvoorbeeld wordt gebruikt na een niertransplantatie), kan uw arts bloedonderzoeken laten uitvoeren om de ciclosporinespiegels te controleren tijdens de behandeling met EPREX.
IJzersupplementen en andere anti-anemische factoren kunnen de werkzaamheid van EPREX verhogen.Uw arts zal beslissen of u ze moet gebruiken.
In geval van ziekenhuisopname of medisch onderzoek, gelieve te informeren dat u behandeld wordt met EPREX. Dit kan andere behandelingen of testresultaten beïnvloeden.
Waarschuwingen Het is belangrijk om te weten dat:
Zwangerschap en borstvoeding
Het is belangrijk om het uw arts te vertellen als een van de volgende situaties op u van toepassing is. U kunt EPREX nog steeds gebruiken, maar bespreek dit eerst met uw arts.
- Als u zwanger bent, of denkt te zijn.
- Als u borstvoeding geeft.
Dosis, wijze en tijdstip van toediening Hoe Eprex te gebruiken: Dosering
Gebruik dit geneesmiddel altijd precies volgens de instructies van uw arts. Raadpleeg uw arts als u niet zeker bent.
Uw arts heeft op basis van uw bloedonderzoek vastgesteld dat u EPREX nodig heeft.
EPREX kan worden toegediend via een injectie:
- In een ader of buis in een ader (intraveneus)
- Onder de huid (subcutaan)
Uw arts zal beslissen hoe EPREX moet worden toegediend. Injecties worden meestal gegeven door een arts, verpleegkundige of zorgverlener; sommige mensen kunnen leren het geneesmiddel zelf subcutaan toe te dienen: zie Instructies voor het zelf injecteren van EPREX.
- Eprex mag niet worden gebruikt:
- na de vervaldatum vermeld op het etiket of de buitenverpakking
- als u weet of denkt dat het geneesmiddel per ongeluk is ingevroren of ja
- en er was een koelkaststoring
De dosis EPREX is gebaseerd op uw lichaamsgewicht in kilogrammen en zal door uw arts worden gekozen afhankelijk van de oorzaak van de bloedarmoede.
Uw arts zal uw bloeddruk regelmatig controleren tijdens de behandeling met EPREX.
Patiënten met nierinsufficiëntie
- Uw hemoglobinewaarde blijft tussen 10 en 12 g/dl, omdat hogere hemoglobinewaarden het risico op trombose en overlijden kunnen vergroten.
- De gebruikelijke startdosering van EPREX voor volwassenen of kinderen is 50 Internationale Eenheden (IE) per kilogram (/kg) lichaamsgewicht, 3 maal per week toegediend.
- Bij patiënten die peritoneale dialyse ondergaan, kan de toediening tweemaal per week worden gedaan.
- EPREX wordt intraveneus (ader of buis in een ader) toegediend aan volwassenen en kinderen. Wanneer de intraveneuze route (ader of buis in een ader) niet beschikbaar is, kan uw arts beslissen of EPREX onder de huid (subcutaan) moet worden geïnjecteerd. Inclusief dialysepatiënten en patiënten die nog niet gedialyseerd worden.
- Uw arts zal regelmatig bloedonderzoek laten doen om te controleren of uw bloedarmoede reageert, en kan de dosis aanpassen, gewoonlijk niet vaker dan eens per 4 weken.
- Zodra de anemie is gecorrigeerd, zal uw arts uw bloedonderzoek regelmatig blijven controleren.De dosis EPREX en de toedieningsfrequentie kunnen verder worden aangepast om de respons op de behandeling te behouden.
- Als u wordt behandeld met EPREX met langere doseringsintervallen (meer dan eenmaal per week), kunt u uw hemoglobinegehalte mogelijk niet voldoende op peil houden en moet u mogelijk uw EPREX-dosis of de toedieningsfrequentie verhogen.
- Om de behandeling effectiever te maken, kunnen ijzersupplementen voor en tijdens de behandeling met EPREX nuttig voor u zijn.
- Als u hemodialyse ondergaat wanneer u begint met de behandeling met EPREX, moet uw dialyseregime mogelijk worden aangepast. De dokter zal beslissen.
Volwassen patiënten die chemotherapie krijgen
- Uw arts kan de behandeling met EPREX starten als uw hemoglobinegehalte 10 g/dl of minder is.
- Uw hemoglobinewaarde blijft tussen 10 en 12 g/dl, omdat hogere hemoglobinewaarden het risico op trombose en overlijden kunnen vergroten.
- De startdosering is 150 IE per kilogram lichaamsgewicht 3 keer per week, of 450 IE per kilogram lichaamsgewicht eenmaal per week.
- EPREX wordt toegediend via subcutane injectie.
- Uw arts zal regelmatig bloedonderzoeken ondergaan en kan de dosis aanpassen, afhankelijk van uw reactie op de behandeling met EPREX.
- Om de behandeling effectiever te maken, kan voor en tijdens de behandeling met EPREX een ijzersupplement nuttig voor u zijn.
- De behandeling met EPREX wordt gewoonlijk gedurende 1 maand na het einde van de chemotherapie voortgezet.
Volwassen patiënten die hun eigen bloed deponeren
- De gebruikelijke dosering is 600 IE per kilogram lichaamsgewicht 2 maal per week.
- EPREX wordt toegediend in een ader onmiddellijk na het afzetten van bloed gedurende 3 weken voorafgaand aan de operatie.
- Om de behandeling effectiever te maken, kan voor en tijdens de behandeling met EPREX een ijzersupplement nuttig voor u zijn.
Volwassen patiënten die in aanmerking komen voor grote orthopedische chirurgie
- De aanbevolen dosering is eenmaal per week 600 IE per kilogram lichaamsgewicht.
- EPREX wordt wekelijks toegediend via subcutane injectie gedurende 3 weken voorafgaand aan de operatie en op de dag van de operatie.
- Als het nodig is om de tijd voor de operatie te verkorten, krijgt u de dagelijkse dosis van 300 IE/kg in de 10 dagen voor de operatie, op de dag van de operatie en in de 4 dagen na de operatie.
- Als uit het bloedonderzoek voor de operatie te hoge hemoglobinewaarden blijken, wordt de behandeling stopgezet.
- Om de behandeling effectiever te maken, kunnen ijzersupplementen voor en tijdens de behandeling met EPREX nuttig voor u zijn.
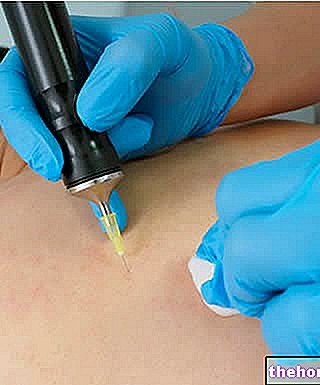
Instructies voor het zelf injecteren van EPREX
- Aan het begin van de behandeling wordt EPREX gewoonlijk toegediend door een arts of verpleegkundige.Daarna kan uw arts u (of uw verzorger) voorstellen om te leren hoe u (subcutaan) moet injecteren.
- Probeer niet uzelf te injecteren als uw arts of verpleegkundige u niet heeft verteld hoe.
- Volg altijd de instructies van uw arts of verpleegkundige op.
- Gebruik EPREX alleen als het correct is bewaard - zie de rubriek Hoe bewaart u EPREX
- Haal de Eprex-spuit voor gebruik uit de koelkast en laat deze op kamertemperatuur komen, dit duurt meestal 15-30 minuten.
Trek een enkele dosis EPREX op uit elke voorgevulde spuit.Wanneer EPREX onder de huid (subcutaan) wordt toegediend, is het volume normaal gesproken niet groter dan 1 milliliter (1 ml) voor een enkele injectie.
EPREX moet alleen worden toegediend en niet worden gemengd met andere injectievloeistoffen.
Schud de EPREX voorgevulde spuiten niet. Langdurig krachtig schudden kan het product beschadigen.Gebruik het product niet als het krachtig geschud is.
Hoe u uzelf kunt injecteren met de voorgevulde spuiten
De voorgevulde spuiten zijn uitgerust met een naaldbeveiliging, PROTECS™, om het risico op het blijven steken van de naald na gebruik te voorkomen. Dit staat op de verpakking aangegeven.
- Haal de voorgevulde spuit voor gebruik uit de koelkast. De vloeistof moet op kamertemperatuur komen. Verwijder de beschermkap niet van de naald terwijl u wacht tot deze op kamertemperatuur is.
- Controleer de voorgevulde spuit om er zeker van te zijn dat het de juiste dosis is, dat de houdbaarheidsdatum niet is verstreken, dat deze niet is beschadigd en dat de vloeistof helder en niet bevroren is.
- Kies de injectieplaats. De meest geschikte plaatsen voor injectie zijn het bovenbeen en de buik, behalve het gebied rond de navel. Verander elke keer van injectieplaats.
- Om handen te wassen. Gebruik een antiseptisch doekje om de injectieplaats te desinfecteren.
- Houd de voorgevulde spuit vast bij de spuitcilinder met de afgedekte naald naar boven gericht.
- Niet vasthouden aan de plunjerkop, de plunjer, de naaldbeschermervleugel of de naaldbeschermerdop.
- Trek de plunjer in geen geval terug
- Verwijder de naalddop van de voorgevulde spuit pas als u klaar bent om EPREX . toe te dienen
- Verwijder de beschermkap van de naald van de spuit door deze bij het lichaam vast te houden en aan de dop te trekken zonder hem te draaien.Druk niet op de zuiger, raak de naald niet aan en schud de spuit niet. - Raak de activeringsclips van het veiligheidsapparaat niet aan om vroegtijdige naaldbescherming met de naaldbeschermingsflap te voorkomen
- Til de huid tussen duim en wijsvinger op zonder er teveel in te knijpen.
- Duw de naald helemaal naar binnen. Uw arts of verpleegkundige heeft u laten zien hoe.
- Duw met uw duim op de zuiger totdat de volledige hoeveelheid vloeistof die overeenkomt met de juiste dosis is geïnjecteerd. Duw langzaam en gelijkmatig, terwijl u de huid omhoog houdt. De PROTECS ™ naaldbeveiliging wordt pas geactiveerd als de injectie is toegediend. volledige dosis. Mogelijk hoort u een "klik" wanneer de PROTECS™ naaldbeveiliging is geactiveerd.
- Wanneer de zuiger het einde van zijn slag heeft bereikt, trekt u de naald eruit en laat u de huid los.
- Verwijder langzaam uw duim van de zuiger zodat de naald volledig door de veiligheidsvoorziening kan worden bedekt.
- Aan het einde van de injectie kan er een kleine hoeveelheid bloed op de injectieplaats zijn.Dit is normaal.U kunt de injectieplaats desinfecteren door enkele seconden op het antiseptische kussentje te drukken.
- Gooi gebruikte spuiten weg in een veilige container - zie rubriek Hoe bewaart u Eprex
Overdosering Wat te doen als u te veel Eprex heeft ingenomen
Wat u moet doen als u meer van EPREX heeft gebruikt dan u zou mogen
Vertel het uw arts of verpleegkundige direct als u denkt dat u te veel EPREX heeft gebruikt.Bijwerkingen van overdosering zijn onwaarschijnlijk.
Wat u moet doen wanneer u bent vergeten EPREX te injecteren
Voer de volgende injectie uit zodra u eraan denkt.Als de volgende injectie binnen een dag valt, sla dan de gemiste dosis over en ga verder met het gebruikelijke schema. Verdubbel de injecties niet.
Als u een patiënt bent met hepatitis C en interferon en ribavirine krijgt
U dient uw arts te raadplegen omdat de combinatie van epoëtine alfa met interferon en ribavirine in zeldzame gevallen heeft geleid tot verlies van effect en de ontwikkeling van een ernstige vorm van bloedarmoede, pure rode-celaplasie (PRCA) genaamd.EPREX is niet goedgekeurd voor de behandeling van bloedarmoede geassocieerd met hepatitis C.
Als u nog vragen heeft over het gebruik van dit product, neem dan contact op met uw arts, verpleegkundige of apotheker.
Bijwerkingen Wat zijn de bijwerkingen van Eprex
Zoals alle geneesmiddelen kan EPREX bijwerkingen hebben, al krijgt niet iedereen daarmee te maken. Vertel het uw arts of verpleegkundige meteen als een van de onderstaande effecten optreedt.
Zeer vaak voorkomende bijwerkingen
Ze komen voor bij meer dan 1 op de 10 patiënten die EPREX gebruiken
- Diarree
- Ziek in de maag voelen
- hij kokhalsde
- Koorts
- Verstopping van de luchtwegen, zoals verstopte neus en keelpijn, is gemeld bij patiënten met een nierziekte die nog geen dialyse ondergaan.
Vaak voorkomende bijwerkingen
Deze komen voor bij maximaal 1 op de 10 patiënten die EPREX gebruiken
- Verhoging van de bloeddrukwaarden. Hoofdpijn (vooral als deze plotseling, acuut en lijkt op migraine) of verwardheid of toevallen Dit kunnen tekenen zijn van een plotselinge stijging van de bloeddruk, die onmiddellijke behandeling vereist De stijging van de bloeddruk kan behandeling vereisen met medicijnen (of een dosisaanpassing van medicijnen die u al gebruikt voor hoge bloeddruk).
- Bloedstolsels (inclusief diepe veneuze trombose en embolie) die mogelijk dringend moeten worden behandeld. Symptomen kunnen zijn pijn op de borst, piepende ademhaling, pijnlijke zwelling van de onderste ledematen en roodheid, meestal in de benen.
- hoest.
- Huidirritatie die kan worden veroorzaakt door een allergische reactie.
- Bot- of spierpijn.
- Griepachtig syndroom, zoals hoofdpijn, pijn in de gewrichten, gevoel van zwakte, koude rillingen, vermoeidheid en duizeligheid. Deze reacties komen vaker voor bij het begin van de behandeling.Als u deze symptomen ervaart tijdens het injecteren in een ader, kan een langzamere toediening helpen ze in de toekomst te voorkomen.
- Roodheid, branderig gevoel en pijn op de injectieplaats.
- Zwelling in de enkels, voeten of vingers.
Soms voorkomende bijwerkingen
Deze komen voor bij maximaal 1 op de 100 patiënten die EPREX gebruiken
- Hoge kaliumspiegels in het bloed die een abnormaal hartritme kunnen veroorzaken (dit is een zeer vaak voorkomende bijwerking bij dialysepatiënten).
- stuiptrekkingen
- Verstopping van de neus of luchtwegen
Zeer zeldzame bijwerkingen
Deze komen voor bij maximaal 1 op de 10.000 patiënten die EPREX gebruiken
- Symptomen van Pure Red Cell Aplasia (PRCA). PRCA betekent onvermogen om voldoende rode bloedcellen in het beenmerg aan te maken. PRCA kan plotselinge en ernstige bloedarmoede veroorzaken, waarvan de symptomen zijn:
- ongebruikelijke vermoeidheid,
- gevoel van duizeligheid,
- ademloosheid.
PRCA is zeer zelden gevonden, vooral bij patiënten met een nierziekte na maanden of jaren behandeling met EPREX en andere stoffen die de aanmaak van rode bloedcellen stimuleren. Vooral aan het begin van de behandeling kan een verhoging van het aantal kleine bloedcellen (bloedplaatjes) optreden, die normaal betrokken zijn bij de vorming van bloedstolsels, bloedplaatjes genaamd.
Als u hemodialyse ondergaat:
- In de dialyseshunt kunnen zich bloedstolsels (trombose) vormen. Dit kan gemakkelijker gebeuren als u een lage bloeddruk (hypotensie) heeft of als u problemen heeft met uw fistel.
- Er kunnen zich ook bloedstolsels vormen in het hemodialysesysteem Uw arts kan besluiten om de dosis heparine tijdens de dialyse te verhogen.
Vertel het uw arts of verpleegkundige onmiddellijk als u een van deze effecten opmerkt, of als u andere effecten bemerkt tijdens het gebruik van EPREX.
Als een van de bijwerkingen ernstig wordt of als er bij u een bijwerking optreedt die niet in deze bijsluiter is vermeld, raadpleeg dan uw arts, verpleegkundige of apotheker.
Voor degenen die aan sport doen: het gebruik van het middel zonder therapeutische noodzaak is doping en kan in ieder geval positieve antidopingtesten opleveren.
Vervaldatum en retentie
Buiten het zicht en bereik van kinderen houden.
Gebruik dit geneesmiddel niet meer na de uiterste houdbaarheidsdatum. Die is te vinden op de doos en het etiket na de letters EXP. De uiterste houdbaarheidsdatum is de laatste dag van de aangegeven maand.
EPREX moet in de koelkast worden bewaard (2°C - 8°C).
EPREX kan uit de koelkast worden gehaald en maximaal 3 dagen bij kamertemperatuur (beneden 25°C) worden bewaard. Zodra de voorgevulde spuit uit de koelkast is gehaald en op kamertemperatuur is (beneden 25°C), moet deze binnen 3 dagen worden gebruikt of worden weggegooid.
Het mag niet worden ingevroren of geschud
Bewaren in de originele verpakking om ze tegen licht te beschermen.
Gebruik dit geneesmiddel niet als de verzegeling is verbroken of als de oplossing verkleurd is of als er zwevende deeltjes worden waargenomen. Als deze omstandigheden worden waargenomen, gooi het geneesmiddel dan weg
Geneesmiddelen dienen niet weggegooid te worden via het afvalwater of met huishoudelijk afval. Vraag uw apotheker wat u met geneesmiddelen moet doen die u niet meer gebruikt. Dit helpt het milieu te beschermen.
Inhoud van de verpakking en andere informatie
Wat bevat Eprex
Het werkzame bestanddeel is: Epoëtine alfa (zie voor de hoeveelheid onderstaande tabel).
De andere stoffen in dit middel zijn: Polysorbaat 80, natriumchloride, monobasisch natriumfosfaatdihydraat, dibasisch natriumfosfaatdihydraat, glycine en water voor injecties. Dit geneesmiddel bevat minder dan 1 mmol natrium (23 mg) per dosis en is dus in wezen natriumvrij.
Hoe ziet EPREX eruit en hoeveel zit er in een verpakking?
EPREX is een oplossing voor injectie in voorgevulde spuiten. De voorgevulde spuiten zijn voorzien van een PROTECS™ naaldbeveiliging (zie onderstaande tabel) EPREX is een heldere en kleurloze oplossing.
Mogelijk worden niet alle verpakkingsgrootten in de handel gebracht
Bron Bijsluiter: AIFA (Italiaans Geneesmiddelenbureau). Inhoud gepubliceerd in januari 2016. De aanwezige informatie is mogelijk niet up-to-date.
Om toegang te hebben tot de meest actuele versie, is het raadzaam om de website van AIFA (Italian Medicines Agency) te bezoeken. Disclaimer en nuttige informatie.
01.0 NAAM VAN HET GENEESMIDDEL
EPREX 10000 IE/ML OPLOSSING VOOR INJECTIE IN VOORGEVULDE SPUIT
02.0 KWALITATIEVE EN KWANTITATIEVE SAMENSTELLING
Epoëtine alfa 10.000 IE / ml (84,0 mcg per ml) geproduceerd door middel van recombinant-DNA-technologie in ovariumcellen van Chinese hamsters (CHO).
Eén voorgevulde spuit van 0,3 ml bevat 3000 IE (25,2 mcg) epoëtine alfa
Eén voorgevulde spuit van 0,4 ml bevat 4.000 IE (33,6 mcg) epoëtine alfa
Eén voorgevulde spuit van 0,5 ml bevat 5.000 IE (42,0 mcg) epoëtine alfa
Eén voorgevulde spuit van 0,6 ml bevat 6.000 IE (50,4 microgram) epoëtine alfa
Eén voorgevulde spuit van 0,8 ml bevat 8.000 IE (67,2 mcg) epoëtine alfa
Eén voorgevulde spuit van 1,0 ml bevat 10.000 IE (84,0 mcg) epoëtine alfa
Dit geneesmiddel bevat minder dan 1 mmol (23 mg) natrium per dosis, dwz het is in wezen "natriumvrij".
Voor de volledige lijst van hulpstoffen, zie rubriek 6.1
03.0 FARMACEUTISCHE VORM
Oplossing voor injectie in voorgevulde spuiten
Heldere, kleurloze oplossing
04.0 KLINISCHE INFORMATIE
04.1 Therapeutische indicaties
EPREX is geïndiceerd voor de behandeling van symptomatische anemie geassocieerd met chronisch nierfalen (CRI):
• bij volwassen en pediatrische patiënten van 1 - 18 jaar die hemodialyse ondergaan en bij volwassen patiënten die peritoneale dialyse ondergaan.
• bij volwassen patiënten met nierinsufficiëntie die nog geen dialyse ondergaan voor de behandeling van ernstige anemie van renale oorsprong die gepaard gaat met klinische symptomen bij patiënten.
EPREX is geïndiceerd bij volwassen patiënten die chemotherapie krijgen voor solide tumoren, maligne lymfoom of multipel myeloom en die een risico lopen op transfusie, zoals blijkt uit de algemene toestand van de patiënt (cardiovasculaire situatie, reeds bestaande anemie bij aanvang van chemotherapie) voor de behandeling van anemie en vermindering van van de behoefte aan transfusie.
EPREX is geïndiceerd bij volwassen patiënten die deelnemen aan een predonatieprogramma om de hoeveelheid autoloog bloed te verhogen Behandeling is alleen geïndiceerd bij patiënten met matige anemie (hemoglobineconcentratie in het bereik van Hb 10-13 g/dl [6, 2-8,1] mmol/l], geen ijzertekort) als er geen of onvoldoende bloedopslagprocedures beschikbaar zijn in het geval van een grote electieve operatie waarbij een grote hoeveelheid bloed nodig is (4 of meer eenheden per vrouw of 5 of meer eenheden voor mannen).
EPREX is geïndiceerd bij volwassen patiënten die voorafgaand aan een grote electieve orthopedische operatie geen ijzertekort hebben en waarvan wordt aangenomen dat ze een hoog risico lopen op transfusiecomplicaties, om de blootstelling aan allogene bloedtransfusies te verminderen. Het gebruik moet worden beperkt tot patiënten met matige anemie ( hemoglobineconcentratie in het bereik van Hb10-13 g / dl), voor wie geen autoloog bloedpredonatieprogramma beschikbaar is en voor wie matig bloedverlies wordt verwacht (van 900 tot 1800 ml).
04.2 Dosering en wijze van toediening
Dosering
Voordat de behandeling met epoëtine alfa wordt gestart en wanneer wordt besloten de dosis te verhogen, alle andere oorzaken van bloedarmoede (ijzer-, folaat- of vitamine B12-tekort, aluminiumintoxicatie, infecties of ontsteking, bloedverlies, hemolyse en beenmergfibrose van welke oorsprong dan ook). Om een optimale respons op epoëtine alfa te garanderen, moet worden gezorgd voor voldoende ijzeropslag en moet zo nodig ijzersuppletie worden toegediend (zie rubriek 4.4).
Behandeling van symptomatische anemie bij volwassen patiënten met chronisch nierfalen (CRI)
Symptomen en gevolgen van bloedarmoede kunnen variëren afhankelijk van geslacht, leeftijd en aanhoudende comorbiditeiten; evaluatie door de arts van de klinische toestand van de individuele patiënt is noodzakelijk.
De gewenste hemoglobineconcentratie ligt tussen 10 g/dl en 12 g/dl (6,2 tot 7,5 mmol/l). EPREX moet zodanig worden toegediend dat een hemoglobineconcentratie wordt bereikt die niet hoger is dan 12 g/dl (7,5 mmol/l). Een hemoglobinestijging van meer dan 2 g/dl (1,25 mmol/l) over een periode van vier weken moet worden vermeden. Als dit gebeurt, moet een passende dosisaanpassing worden gemaakt.
Vanwege de variabiliteit binnen de patiënt kunnen bij een patiënt af en toe hemoglobinewaarden boven en onder de gewenste hemoglobineconcentratie worden waargenomen. Deze variabiliteit moet worden beheerd door middel van dosisaanpassingen, met inachtneming van het hemoglobineconcentratiebereik tussen 10 g / dl (6,2 mmol / l) en 12 g / dl (7,5 mmol / l).
Een hemoglobinegehalte dat constant boven de 12 g/dl (7,5 mmol) ligt, moet worden vermeden. Als het hemoglobinegehalte meer dan 2 g/dl (1,25 mmol/l) per maand stijgt of als het hemoglobinegehalte constant 12 g/dl (7,5 mmol) overschrijdt, moet de dosis EPREX met 25% worden verlaagd. (8,1 mmol / l), onderbreek de therapie totdat deze terugkeert naar 12 g / dl (7,5 mmol / l) en ga dan verder met EPREX met doses die 25% lager zijn dan de vorige.
Patiënten moeten nauwlettend worden gecontroleerd om er zeker van te zijn dat de laagst toegestane effectieve dosis EPREX wordt gebruikt om een adequate beheersing van bloedarmoede en gerelateerde symptomen te garanderen door een hemoglobineconcentratie onder of gelijk aan 12 g/dl (7, 5 mmol/l) te handhaven.
Voorzichtigheid is geboden bij het verhogen van de ESA-dosis bij patiënten met chronisch nierfalen.Voor patiënten met een slechte hemoglobinerespons op ESA's moet worden gezocht naar een alternatieve oorzaak van de slechte respons (zie rubrieken 4.4 en 5.1).
De EPREX-behandeling is verdeeld in twee fasen: de correctiefase en de onderhoudsfase.
Volwassen patiënten die hemodialyse ondergaan
Bij hemodialysepatiënten bij wie veneuze toegang direct beschikbaar is, verdient het gebruik van de intraveneuze toedieningsweg de voorkeur.
Correctie fase :
De startdosering is 3 maal per week 50 IE/kg gewicht.
Verhoog of verlaag indien nodig de dosis met 25 IE/kg (3 keer per week) tot de gewenste hemoglobineconcentratie in het bereik van 10g/dl tot 12g/dl (6,2 tot 7,5 mmol/l) (dit moet geleidelijk tussenpozen van minimaal vier weken).
- Onderhoudsfase :
De aanbevolen totale wekelijkse dosis ligt tussen 75 IE/kg en 300 IE/kg.
Er moet een passende dosisaanpassing worden gedaan om de hemoglobinewaarden binnen de gewenste hemoglobineconcentratie tussen 10 g/dl en 12 g/dl (6,2 tot 7,5 mmol/l) te houden.
Patiënten met een zeer laag aanvankelijk hemoglobinegehalte (8 g / dl of> 5 mmol / l).
Volwassen patiënten met nierinsufficiëntie die nog geen dialyse ondergaan
Bij patiënten bij wie veneuze toegang niet direct beschikbaar is, kan EPREX subcutaan worden toegediend.
Correctie fase
De startdosering is 50 IE/kg gewicht, 3 keer per week, indien nodig gevolgd door een dosisverhoging van 25 IE/kg (3 keer per week) totdat de gewenste hemoglobineconcentratie is bereikt (dit dient geleidelijk te gebeuren. tussenpozen van minimaal vier weken).
Onderhoudsfase
Tijdens de onderhoudsfase kan EPREX ofwel 3 keer per week worden toegediend en, in geval van subcutane toediening, één keer per week of één keer per twee weken.
De dosis en doseringsintervallen moeten correct worden aangepast om de hemoglobinewaarden op het gewenste niveau te houden: Hb tussen 10 en 12 g/dl (6,2-7,5 mmol/l). Het verlengen van het doseringsinterval kan een dosisverhoging vereisen.
De maximale dosis mag niet hoger zijn dan 150 IE/kg 3 keer per week, 240 IE/kg (tot maximaal 20.000 IE) eenmaal per week of 480 IE/kg (tot maximaal 40.000 IE) eenmaal per 2 weken.
Volwassen patiënten die peritoneale dialyse ondergaan
Bij patiënten bij wie veneuze toegang niet direct beschikbaar is, kan EPREX subcutaan worden toegediend.
Correctie fase
De startdosering is 50 IE/kg, tweemaal per week.
Onderhoudsfase
De aanbevolen onderhoudsdosering ligt tussen 25 IE/kg en 50 IE/kg, tweemaal per week, verdeeld over 2 gelijke toedieningen.
Er moet een passende dosisaanpassing worden gedaan om de hemoglobinewaarden op het gewenste niveau te houden: hemoglobine tussen 10 g / dl en 12 g / dl (6,2-7,5 mmol / l).
Behandeling van volwassen patiënten met door chemotherapie geïnduceerde anemie
De symptomen en gevolgen van bloedarmoede kunnen variëren afhankelijk van leeftijd, geslacht en de algemene toestand van de ziekte; de beoordeling door de arts van de klinische toestand van de individuele patiënt is noodzakelijk.
EPREX moet worden toegediend aan anemische patiënten, bijv. hemoglobineconcentratie ≤10 g / dl (6,2 mmol / l).
De startdosering is 150 IE/kg, subcutaan toegediend, 3 keer per week.
Als alternatief kan EPREX subcutaan worden toegediend in een startdosering van 450 IE/kg eenmaal per week.
Er moet een passende dosisaanpassing worden gedaan om de hemoglobinewaarden op het gewenste niveau te houden: hemoglobine tussen 10 g / dl en 12 g / dl (6,2-7,5 mmol / l).
Vanwege de variabiliteit binnen de patiënt kunnen bij een patiënt af en toe hemoglobinewaarden boven en onder de gewenste hemoglobineconcentratie worden waargenomen. Deze variabiliteit moet worden beheerd door middel van dosisaanpassingen, waarbij het gewenste hemoglobineconcentratiebereik tussen 10 g / dl (6,2 mmol / l) en 12 g / dl (7,5 mmol / l) wordt gerespecteerd.
Een hemoglobineconcentratie die constant boven de 12 g/dl (7,5 mmol) ligt, moet worden vermeden. Hieronder vindt u instructies voor een juiste dosisaanpassing als hemoglobine een concentratie van meer dan 12 g/dl (7,5 mmol) bereikt.
Als na 4 weken behandeling de hemoglobineconcentratie is gestegen met ten minste 1 g/dl (0,62 mmol/l), of als het aantal reticulocyten is toegenomen met ≥ 40.000 cellen/mcl vanaf de uitgangswaarde, moet de dosis op 150 IE/mcl blijven. kg 3 keer per week of 450 IE / kg eenmaal per week.
Als de toename van de hemoglobineconcentratie is
Als de verhoging van de hemoglobineconcentratie was
Dosisaanpassing om hemoglobineconcentraties tussen 10 g / dl - 12 g / dl . te houden
Als de hemoglobineconcentratie meer dan 2 g/dl (1,25 mmol/l) per maand stijgt, of als de hemoglobine 12 g/dl (7,5 mmol/l) overschrijdt, verlaag dan de dosis EPREX, met ongeveer 25-50%.
Als de hemoglobineconcentratie hoger is dan 13 g / dl (8,1 mmol / l), stop dan met de therapie totdat de concentratie onder 12 g / dl (7,5 mmol / l) daalt en hervat vervolgens de behandeling met EPREX in een dosis die 25% lager is dan de vorige dosis.
De aanbevolen dosering wordt beschreven in het volgende diagram:
Patiënten moeten nauwlettend worden gecontroleerd om er zeker van te zijn dat de laagst goedgekeurde dosis erytropoëse-stimulerende middelen (ESA) wordt gebruikt om de symptomen van anemie adequaat onder controle te houden.
De EPREX-therapie moet gedurende één maand na het einde van de chemotherapie worden voortgezet.
Behandeling van volwassen patiënten die in aanmerking komen voor een operatie die deel uitmaakt van een predonatieprogramma voor autoloog bloed
Licht anemische patiënten (hematocriet tussen 33-39%) die een pre-depositie van 4 of meer eenheden bloed nodig hebben, dienen te worden behandeld met 600 IE/kg EPREX intraveneus, tweemaal per week, gedurende de 3 weken voorafgaand aan de operatie.
EPREX moet worden toegediend na voltooiing van de bloeddonatieprocedure.
Behandeling van volwassen patiënten die in aanmerking komen voor grote electieve orthopedische chirurgie
De aanbevolen dosis is 600 IE/kg EPREX subcutaan toegediend, eenmaal per week gedurende de drie weken voorafgaand aan de operatie (-21 dagen, -14 dagen en -7 dagen) en op de dag van de operatie.
Als er een medische noodzaak is om de wachttijd vóór de operatie te verkorten tot minder dan drie weken, dient de dosis van 300 IE/kg EPREX dagelijks gedurende 10 opeenvolgende dagen subcutaan te worden toegediend, vóór de operatie, op de dag van de operatie en in de 4 dagen die er direct op volgen.
Als hemoglobine tijdens de preoperatieve periode 15 g/dl of meer bereikt, moet de toediening van EPREX worden gestaakt en mogen geen verdere doses worden toegediend.
Pediatrische populatie
Behandeling van symptomatische anemie bij patiënten met chronisch nierfalen die hemodialyse ondergaan
Symptomen en gevolgen van bloedarmoede kunnen variëren afhankelijk van leeftijd, geslacht en aanhoudende comorbiditeiten; evaluatie door de arts van de klinische toestand van de individuele patiënt is noodzakelijk.
Bij pediatrische patiënten ligt de gewenste hemoglobineconcentratie tussen 9,5 g/dl en 11 g/dl (5,9 tot 6,8 mmol/l). EPREX moet zodanig worden toegediend dat een hemoglobineconcentratie wordt bereikt die niet hoger is dan 11 g/dl (6,8 mmol/l). Een hemoglobinestijging van meer dan 2 g/dl (1,25 mmol/l) over een periode van vier weken moet worden vermeden. Als dit gebeurt, moet een geschikte dosisaanpassing worden gemaakt.
Patiënten moeten nauwlettend worden gecontroleerd om er zeker van te zijn dat de laagst toegestane dosis EPREX wordt gebruikt om een adequate controle van anemie en gerelateerde symptomen te verzekeren.
De EPREX-behandeling is verdeeld in twee fasen: de correctiefase en de onderhoudsfase.
Bij pediatrische patiënten die hemodialyse ondergaan en waarbij intraveneuze toegang al beschikbaar is, verdient intraveneuze toediening de voorkeur.
Correctiefase:
De startdosering is 50 IE/kg gewicht intraveneus, 3 keer per week.
Verhoog of verlaag indien nodig de dosis met 25 IE / kg (3 keer per week) tot de gewenste hemoglobineconcentratie in het bereik van 9,5 g / dl tot 11 g / dl (5,9 tot 6, 8 mmol / l) (dit zou moeten gebeuren geleidelijk met tussenpozen van ten minste vier weken).
Onderhoudsfase :
Er moet een passende dosisaanpassing worden gedaan om de hemoglobinewaarden binnen de gewenste hemoglobineconcentratie tussen 9,5 g/dl en 11 g/dl (5,9 tot 6,8 mmol/l) te houden.
Over het algemeen hebben kinderen die minder dan 30 kg wegen een hogere onderhoudsdosering nodig dan kinderen die meer dan 30 kg wegen en volwassenen.
Pediatrische patiënten met een zeer laag aanvankelijk hemoglobinegehalte (6,8 g / dL of> 4,25 mmol / L).
Behandeling van pediatrische patiënten met door chemotherapie geïnduceerde bloedarmoede.
De veiligheid en werkzaamheid van EPREX bij pediatrische patiënten die chemotherapie krijgen, zijn niet vastgesteld.
Behandeling van pediatrische chirurgische patiënten die deelnemen aan een autoloog predonatieprogramma
De veiligheid en werkzaamheid van EPREX bij pediatrische patiënten zijn niet vastgesteld.
Er zijn geen gegevens beschikbaar.
Behandeling van pediatrische patiënten die wachten op een grote electieve orthopedische operatie
De veiligheid en werkzaamheid van EPREX bij pediatrische patiënten zijn niet vastgesteld.
Er zijn geen gegevens beschikbaar.
Wijze van toediening
Wees voorzichtig voordat u het geneesmiddel gebruikt of toedient.
Laat de EPREX-spuit voor gebruik rusten totdat deze op kamertemperatuur is, dit duurt meestal tussen de 15 en 30 minuten.
Behandeling van symptomatische anemie bij volwassen patiënten met chronisch nierfalen
Bij patiënten met chronisch nierfalen bij wie intraveneuze toegang normaal beschikbaar is (hemodialysepatiënten) verdient intraveneuze toediening van EPREX de voorkeur.
Waar intraveneuze toegang niet direct beschikbaar is (patiënten die nog geen dialyse ondergaan en patiënten die peritoneale dialyse ondergaan), kan EPREX subcutaan worden toegediend.
Behandeling van volwassen patiënten met door chemotherapie geïnduceerde anemie.
EPREX moet subcutaan worden toegediend.
Behandeling van volwassen chirurgische patiënten die deelnemen aan een autoloog predonatieprogramma
EPREX moet intraveneus worden toegediend.
Behandeling van volwassen patiënten die voor een grote electieve orthopedische ingreep zijn ingepland
EPREX moet subcutaan worden toegediend.
Behandeling van symptomatische anemie bij pediatrische patiënten met chronisch nierfalen die hemodialyse ondergaan
Bij pediatrische patiënten met chronisch nierfalen bij wie intraveneuze toegang al beschikbaar is (hemodialysepatiënten), verdient intraveneuze toediening van EPREX de voorkeur.
Intraveneuze toediening
De toediening moet minstens 1-5 minuten duren, afhankelijk van de totale dosis.
Bij hemodialysepatiënten kan de bolusinjectie tijdens de dialyse worden gegeven via een adequate veneuze toegang in de dialyselijn.Als alternatief kan de injectie worden gegeven aan het einde van de dialyse, via de toegang tot de fistel, gevolgd door toediening van 10 ml fysiologische oplossing om de toegangswegen te spoelen en een bevredigende introductie van het product in de bloedbaan te verzekeren.
Bij patiënten die griepachtige reacties hebben gehad, verdient een langzamere toediening de voorkeur (zie rubriek 4.8).
Dien EPREX niet toe via intraveneuze infusie of in oplossing met andere geneesmiddelen.
Subcutane toediening
Het maximale volume van 1 ml op elke injectieplaats mag in het algemeen niet worden overschreden. Voor grotere volumes moet meer dan één injectieplaats worden gekozen.
Injecties moeten worden gedaan in de ledematen of de voorste buikwand.
In het geval dat de arts van mening is dat de patiënt of verzorger in staat is om EPREX veilig en op de juiste manier subcutaan toe te dienen, moeten instructies voor de juiste dosering en toediening worden verstrekt.
Net als bij andere injecteerbare producten, controleer of er geen deeltjes in de oplossing of kleurvariaties zijn.
04.3 Contra-indicaties
Overgevoeligheid voor de werkzame stof of voor één van de hulpstoffen.
Patiënten die Pure Red Cell Aplasia (PRCA) ontwikkelen na behandeling met een erytropoëtine, mogen niet worden behandeld met EPREX of andere erytropoëtines (zie rubriek 4.4 PRCA).
Ongecontroleerde hypertensie.
Bij patiënten die met EPREX worden behandeld, moeten alle contra-indicaties die verband houden met het programma voor predepositie van autoloog bloed in gedachten worden gehouden.
Het gebruik van EPREX is gecontra-indiceerd bij ernstige vasculaire aandoeningen op coronair, perifeer arterieel, carotis- of cerebraal niveau bij patiënten die in aanmerking komen voor een grote electieve orthopedische ingreep en die geen deel uitmaken van een autoloog predonatieprogramma.Het gebruik is ook gecontra-indiceerd. bij patiënten met recente episodes van een myocardinfarct of andere cerebrovasculaire complicaties.
Patiënten die in aanmerking komen voor een operatie en om welke reden dan ook geen adequate antitrombotische profylaxe kunnen krijgen.
04.4 Bijzondere waarschuwingen en passende voorzorgen bij gebruik
Algemeen
Bij alle patiënten die epoëtine alfa krijgen, moet de bloeddruk nauwlettend worden gecontroleerd en zo nodig worden gecontroleerd. Epoëtine alfa moet met voorzichtigheid worden gebruikt in aanwezigheid van onbehandelde, onvoldoende behandelde of moeilijk onder controle te houden hypertensie. Het kan nodig zijn om een antihypertensieve behandeling te starten of te intensiveren. Als de bloeddruk niet onder controle kan worden gebracht, moet de behandeling met epoëtine alfa worden stopgezet.
Hypertensieve crises met encefalopathie en toevallen, die onmiddellijke medische aandacht en intensieve medische zorg vereisen, zijn opgetreden tijdens de behandeling met epoëtine alfa, zelfs bij patiënten met eerdere normale of lage bloeddruk. Bijzondere aandacht moet worden besteed aan migraine-achtige steken als een mogelijk waarschuwingssignaal (zie rubriek 4.8).
Epoëtine alfa moet met voorzichtigheid worden gebruikt bij patiënten met epilepsie, een voorgeschiedenis van convulsies of medische aandoeningen die verband houden met aanleg voor convulsies, zoals infecties van het centrale zenuwstelsel en hersenmetastasen.
Epoëtine alfa moet met voorzichtigheid worden gebruikt bij patiënten met chronische leverinsufficiëntie.De veiligheid van epoëtine alfa is niet vastgesteld bij patiënten met leverdisfunctie.
Een verhoogde incidentie van vasculaire trombotische voorvallen (VTE's) is waargenomen bij patiënten die ESA's hebben gekregen (zie rubriek 4.8), waaronder veneuze en arteriële trombose en embolie (waaronder sommige met fatale afloop), zoals diepe veneuze trombose, longembolie, retinale trombose en myocardinfarct Daarnaast zijn cerebrovasculaire accidenten (waaronder herseninfarct, hersenbloeding en voorbijgaande ischemische aanvallen) gemeld.
Het risico van deze VTE's moet zorgvuldig worden afgewogen tegen het voordeel van behandeling met epoëtine alfa, met name bij patiënten met reeds bestaande risicofactoren voor VTE, waaronder obesitas en een voorgeschiedenis van VTE (bijv. diepe veneuze trombose, longembolie en cerebrovasculaire accidenten)
De hemoglobinespiegels moeten bij alle patiënten nauwlettend worden gecontroleerd vanwege een mogelijk verhoogd risico op trombo-embolische voorvallen en fatale afloop wanneer patiënten worden behandeld met hemoglobinespiegels boven de aangegeven concentratie.
Tijdens behandeling met epoëtine alfa kan een matige dosisafhankelijke toename van het aantal bloedplaatjes optreden, maar binnen het normale bereik. Dit fenomeen neemt af in de loop van de therapie. Bovendien is trombocytemie boven het normale bereik gemeld Regelmatige controle van de bloedplaatjes tijdens de eerste 8 weken van de behandeling wordt aanbevolen.
Alle mogelijke oorzaken van bloedarmoede (ijzertekort, folaat- of vitamine B12-tekort, aluminiumvergiftiging, infectie of ontsteking, bloedverlies, hemolyse en beenmergfibrose van welke oorsprong dan ook) moeten worden geëvalueerd en behandeld voordat de behandeling met epoëtine alfa wordt gestart en wanneer een beslissing wordt genomen. wordt gedaan om de dosis te verhogen. In de meeste gevallen nemen de serum-ferritinewaarden af terwijl de hematocrietwaarden stijgen.Om een optimale respons op epoëtine alfa te garanderen, moet worden gezorgd voor voldoende ijzeropslag en moeten, indien nodig, ijzersupplementen worden gegeven ( zie paragraaf 4.2):
• Voor patiënten met chronisch nierfalen wordt ijzersuppletie aanbevolen als het ferritinegehalte lager is dan 100 ng/ml (elementair ijzer voor volwassenen 200 tot 300 mg/dag oraal en voor kinderen van 100 tot 200 mg/dag oraal).
• Voor kankerpatiënten wordt ijzersuppletie aanbevolen als de transferrineverzadigingswaarden lager zijn dan 20% (elementair ijzer 200 tot 300 mg/dag oraal).
• Voor patiënten in een autoloog predonatieprogramma dient ijzersuppletie (elementair ijzer 200 mg/dag oraal) enkele weken voor de start van autologe predonatie te worden toegediend om een hoge ijzervoorraad te bereiken voordat de behandeling met epoëtine alfa wordt gestart en tijdens de kuur van therapie met epoëtine alfa.
• Voor patiënten die voor een grote electieve orthopedische ingreep zijn ingepland, dient ijzersuppletie (elementair ijzer 200 mg/dag oraal) te worden toegediend tijdens de behandeling met epoëtine alfa. "ijzersuppletie vóór aanvang van de behandeling met epoëtine alfa om een adequate ijzerreserve te bereiken.
Begin of verergering van porfyrie is zeer zelden waargenomen bij patiënten die met epoëtine alfa werden behandeld.
Epoëtine alfa moet met voorzichtigheid worden gebruikt bij patiënten met porfyrie.
Om de traceerbaarheid van erytropoësestimulerende middelen (ESA) te garanderen, moet de handelsnaam van de toegediende ESA altijd worden geregistreerd of vermeld in het medisch dossier van de patiënt.
Het veranderen van therapie van de ene ESA naar de andere mag alleen worden gedaan onder passend toezicht.
Zuivere rode cel aplasie (PRCA)
Antilichaam-gemedieerde Pure Red Cell Aplasia (PRCA) is na maanden tot jaren gemeld, voornamelijk bij patiënten met chronisch nierfalen die werden behandeld met subcutaan toegediende epoëtine.
Er zijn ook gevallen gemeld bij patiënten met hepatitis C die werden behandeld met interferon en ribavirine in combinatie met ESA. Epoëtine alfa is niet goedgekeurd voor de behandeling van anemie geassocieerd met hepatitis C.
Bij patiënten die een plotseling verlies van werkzaamheid vertonen, gedefinieerd als een verlaging van de hemoglobinewaarden (1 tot 2 g/dl per maand) met een verhoogde behoefte aan transfusies, moet een telling van de reticulocyten worden uitgevoerd en moeten bekende oorzaken worden geëvalueerd. reactie (zoals ijzer-, folaat- en vitamine B12-tekort, aluminiumvergiftiging, infectie of ontsteking, bloedverlies, hemolyse en beenmergfibrose van welke oorsprong dan ook).
Een onevenredige daling van de hemoglobinewaarden en de ontwikkeling van ernstige anemie geassocieerd met een laag aantal reticulocyten moet leiden tot stopzetting van de behandeling met epoëtine alfa en het uitvoeren van een test op de aanwezigheid van anti-erytropoëtine-antilichamen.
Een beenmergonderzoek voor een diagnose van PRCA moet ook worden overwogen.
Er mag geen behandeling met andere ESA's worden gestart vanwege het risico op kruisreacties.
Behandeling van symptomatische anemie bij volwassen en pediatrische patiënten met chronisch nierfalen (CRI):
Bij patiënten met chronisch nierfalen die epoëtine alfa krijgen, moet hun hemoglobinegehalte regelmatig worden gemeten totdat een stabiel niveau is bereikt en daarna periodiek worden gemeten.
Bij patiënten met chronisch nierfalen om het risico op bloeddrukstijgingen te verminderen, moet de hemoglobineconcentratie met ongeveer 1 g/dl/maand (0,62 mmol/l) stijgen en mag niet hoger zijn dan 2 g/dl/maand (1,25 mmol/l).
Bij patiënten met chronische nierinsufficiëntie mag de hemoglobine-onderhoudsconcentratie niet hoger zijn dan de maximale waarde van het hemoglobineconcentratiebereik zoals vermeld in rubriek 4.2 Een verhoogd risico op mortaliteit en ernstige cardiovasculaire voorvallen is waargenomen in klinische onderzoeken waarbij erytropoësestimulerende middelen (ESA's) werden gebruikt. toegediend om een hemoglobineconcentratie van meer dan 12 g/dl (7,5 mmol/ml) te bereiken.
Gecontroleerde klinische onderzoeken hebben geen significant voordeel aangetoond dat kan worden toegeschreven aan de toediening van epoëtinen wanneer de hemoglobineconcentratie het niveau overschrijdt dat nodig is om de symptomen van anemie onder controle te houden en bloedtransfusies te vermijden.
Voorzichtigheid is geboden bij het verhogen van de dosis EPREX bij patiënten met chronisch nierfalen, aangezien hoge cumulatieve doses epoëtine in verband kunnen worden gebracht met een verhoogd risico op mortaliteit, ernstige cardiovasculaire en cerebrovasculaire voorvallen Voor patiënten met een slechte hemoglobinerespons op epoëtine, een alternatief Er moet gezocht worden naar een oorzaak voor deze slechte respons (zie rubrieken 4.2 en 5.1).
Patiënten met chronisch nierfalen die worden behandeld met subcutaan epoëtine alfa, moeten regelmatig worden gecontroleerd op verlies van werkzaamheid, dat wil zeggen afwezigheid of verminderde respons op epoëtine alfa bij patiënten die eerder op de behandeling reageerden. Dit fenomeen wordt gekenmerkt door een aanhoudende daling van de hemoglobinewaarden in vergelijking met een verhoging van de dosis epoëtine alfa (zie rubriek 4.8).
Sommige patiënten die worden behandeld met epoëtine alfa met langere doseringsintervallen (meer dan eenmaal per week) behouden mogelijk niet voldoende hemoglobinewaarden (zie rubriek 5.1) en kunnen een dosisverhoging nodig hebben. De hemoglobinespiegels moeten regelmatig worden gecontroleerd.
Trombose van vasculaire toegangen is opgetreden bij patiënten die hemodialyse ondergaan, vooral bij patiënten met een neiging tot hypotensie en met complicaties van arterioveneuze fistels (bijv. stenose, aneurysma's, enz.) Bij deze patiënten wordt preventieve controle van de vasculaire toegang aanbevolen en profylaxe van trombose bij toediening van bijv. acetylsalicylzuur.
Hyperkaliëmie is waargenomen in geïsoleerde gevallen, hoewel de causaliteit niet is vastgesteld. Serumelektrolyten moeten worden gecontroleerd bij patiënten met chronisch nierfalen. Als verhoogde (of stijgende) serumkaliumspiegels worden waargenomen, moet naast een geschikte behandeling van hyperkaliëmie worden overwogen om de toediening van epoëtine alfa stop te zetten totdat de serumkaliumspiegels gecorrigeerd zijn.
Vaak is tijdens hemodialyse een verhoging van de heparinedosis nodig vanwege de verhoging van de hematocrietwaarde. Als de aanpassing van de heparinedosering niet optimaal is, kan occlusie van de dialysator optreden. Op basis van de tot dusver beschikbare gegevens versnelt de correctie van anemie met epoëtine alfa bij volwassen patiënten met chronisch nierfalen die nog geen dialyse ondergaan de progressie van nierfalen niet.
Behandeling van patiënten met door chemotherapie geïnduceerde anemie
Bij kankerpatiënten die epoëtine alfa krijgen, moet hun hemoglobinegehalte regelmatig worden gemeten totdat een stabiel niveau is bereikt en daarna periodiek worden gemeten.
Erytropoëtines zijn groeifactoren die in wezen de aanmaak van rode bloedcellen stimuleren. Erytropoëtinereceptoren kunnen tot expressie worden gebracht op het oppervlak van een verscheidenheid aan kankercellen. Zoals bij alle groeifactoren bestaat er theoretische bezorgdheid dat erytropoëtines de tumorgroei kunnen stimuleren.
In sommige gecontroleerde klinische onderzoeken is niet aangetoond dat erytropoëtines de algehele overleving verbeteren of het risico op tumorprogressie verminderen bij patiënten met tumor-geassocieerde anemie.
In gecontroleerde klinische onderzoeken heeft het gebruik van epoëtine alfa en andere erytropoëse-stimulerende middelen (ESA's) aangetoond:
• verminderde locoregionale controle bij patiënten met gevorderde hoofd-halskanker die radiotherapie krijgen bij toediening om hemoglobinewaarden boven 14 g/dl (8,7 mmol/l) te bereiken;
• verminderde algehele overleving en verhoogde mortaliteit als gevolg van ziekteprogressie na 4 maanden bij patiënten met gemetastaseerde borstkanker die chemotherapie krijgen bij toediening om hemoglobinewaarden tussen 12-14 g/dl (7,5-8,7 mmol/l) te bereiken;
• verhoogd risico op sterfte bij toediening om hemoglobinewaarden van 12 g/dl (7,5 mmol/l) te bereiken; bij patiënten met actieve neoplasie die geen chemo- en/of radiotherapiebehandeling ondergaan. Behandeling met erytropoësestimulerende middelen (ESA's) is niet geïndiceerd bij deze patiëntenpopulatie.
In het licht van het bovenstaande moet in sommige klinische situaties voor de behandeling van anemie bij kankerpatiënten de voorkeur worden gegeven aan bloedtransfusie. De beslissing om recombinant erytropoëtine toe te dienen moet gebaseerd zijn op een baten-risicobeoordeling met individuele betrokkenheid. om hun specifieke klinische context in overweging te nemen Factoren die tijdens deze beoordeling in overweging moeten worden genomen, moeten het type kanker en de voortgang ervan zijn; de mate van bloedarmoede; levensverwachting; de omgeving waarin de patiënt wordt behandeld; patiëntvoorkeuren (zie rubriek 5.1).
Bij kankerpatiënten die chemotherapie krijgen, moet het interval van 2-3 weken tussen toediening en het verschijnen van ESA-geïnduceerde rode bloedcellen zorgvuldig worden overwogen bij het beoordelen van de geschiktheid van behandeling met epoëtine alfa (patiënten met risico op transfusie).
Volwassen patiënten die in aanmerking komen voor chirurgische ingrepen die deel uitmaken van predonatieprogramma's voor autoloog bloed
Alle waarschuwingen en speciale voorzorgsmaatregelen die verband houden met het autologe bloedpredonatieprogramma, met name routinematige volumevervanging, moeten in acht worden genomen.
Patiënten kandidaten voor grote electieve orthopedische chirurgie
Preoperatief moeten altijd goede bloedbeheerpraktijken worden gevolgd.
Patiënten die in aanmerking komen voor electieve grote orthopedische chirurgie moeten "adequate antitrombotische profylaxe krijgen, aangezien trombotische en vasculaire voorvallen kunnen optreden bij patiënten die een operatie ondergaan, vooral bij patiënten met onderliggende cardiovasculaire aandoeningen. Bovendien moeten speciale voorzorgsmaatregelen worden genomen bij patiënten met aanleg voor de ontwikkeling van diepe veneuze trombose (DVT). Bovendien kan bij patiënten met een "baseline hemoglobine> 13 g / dl, de mogelijkheid dat behandeling met epoëtine alfa gepaard gaat met een verhoogd risico op posttrombotische / vasculaire voorvallen. uitgesloten. Daarom mag epoëtine alfa niet worden gebruikt bij patiënten met een baseline hemoglobine > 13 g/dl.
04.5 Interacties met andere geneesmiddelen en andere vormen van interactie
Er is geen bewijs dat behandeling met epoëtine alfa het metabolisme van andere geneesmiddelen verandert. Geneesmiddelen die de erytropoëse verminderen, kunnen de respons op epoëtine alfa verminderen.
Omdat cyclosporine zich bindt aan rode bloedcellen, kan er een "interactie met dit medicijn zijn.
In geval van gelijktijdige toediening moeten de bloedspiegels van ciclosporine worden gecontroleerd en moet de dosis worden aangepast aan de stijging van de hematocriet.
Er zijn geen in vitro interacties tussen G-CSF, GM-CSF en epoëtine alfa met betrekking tot hematologische differentiatie of proliferatie in vitro van tumorbiopten.
Bij volwassen vrouwelijke patiënten met gemetastaseerde borstkanker had de subcutane gelijktijdige toediening van 40.000 IE/ml epoëtine alfa met trastuzumab 6 mg/kg geen effect op de farmacokinetiek van trastuzumab.
04.6 Zwangerschap en borstvoeding
Zwangerschap
Er zijn geen adequate en goed gecontroleerde onderzoeken bij zwangere vrouwen. Uit dieronderzoek is reproductietoxiciteit gebleken (zie rubriek 5.3). Daarom mag epoëtine alfa alleen tijdens de zwangerschap worden gebruikt als het verwachte voordeel opweegt tegen het mogelijke risico voor de foetus. Het gebruik van epoëtine alfa wordt niet aanbevolen bij zwangere vrouwen die in aanmerking komen voor een operatie die deelneemt aan een autoloog bloedpredonatieprogramma.
Voedertijd
Het is niet bekend of exogeen epoëtine alfa wordt uitgescheiden in de moedermelk. Epoëtine alfa moet met voorzichtigheid worden gebruikt bij vrouwen die borstvoeding geven. De beslissing om de borstvoeding voort te zetten/staakt of om de behandeling met epoëtine alfa voort te zetten/stopt, moet worden genomen met overweging van het voordeel van borstvoeding voor het kind en het voordeel van therapie met epoëtine alfa voor de vrouw.
Het gebruik van epoëtine alfa wordt niet aanbevolen bij vrouwen die borstvoeding geven en die in aanmerking komen voor een operatie die deelneemt aan een autoloog bloedpredonatieprogramma.
Vruchtbaarheid
Er zijn geen onderzoeken die het mogelijke effect van epoëtine alfa op de mannelijke of vrouwelijke vruchtbaarheid evalueren.
04.7 Beïnvloeding van de rijvaardigheid en het vermogen om machines te bedienen
Er is geen onderzoek gedaan naar de rijvaardigheid of het vermogen om machines te bedienen.
04.8 Bijwerkingen
Samenvatting van het veiligheidsprofiel
De meest voorkomende bijwerking tijdens de behandeling met epoëtine alfa is een dosisafhankelijke verhoging van de bloeddruk of verergering van reeds bestaande hypertensie.
Het wordt aanbevolen om de bloeddruktrend te controleren, vooral aan het begin van de therapie (zie rubriek 4.4).
De meest voorkomende bijwerkingen die optraden in klinische onderzoeken met epoëtine alfa zijn diarree, misselijkheid, braken, pyrexie en hoofdpijn. Griepachtige symptomen kunnen voornamelijk aan het begin van de behandeling optreden.
Verstopping van de luchtwegen, waaronder voorvallen van congestie van de bovenste luchtwegen, verstopte neus en rhinoparyngitis, is gemeld in klinische onderzoeken naar dosisbereikverlenging bij volwassen patiënten met nierinsufficiëntie die nog geen dialyse ondergaan.
Een verhoogde incidentie van vasculaire trombotische voorvallen (TVE) is waargenomen bij patiënten die ESA kregen (zie rubriek 4.4).
Tabel met bijwerkingen
Van een totaal van 3.262 patiënten in 23 gerandomiseerde, dubbelblinde, placebo- of standaard-of-care gecontroleerde klinische onderzoeken, werd het algehele veiligheidsprofiel van EPREX geëvalueerd bij 1.992 anemische patiënten. In 4 onderzoeken naar chronisch nierfalen werden 228 ICR-patiënten behandeld met epoëtine alfa opgenomen (2 onderzoeken naar predialyse [N = 131 ICR-blootgestelde patiënten] en 2 naar dialyse [N = 97 ICR-blootgestelde patiënten]); 1404 kankerpatiënten blootgesteld in 16 studies over anemie veroorzaakt door chemotherapie 147 patiënten blootgesteld in 2 studies voor predonatie van autoloog bloed en 213 patiënten blootgesteld in 1 studie in de perioperatieve periode Bijwerkingen gemeld bij ≥1% van de patiënten behandeld met epoëtine alfa in deze klinische onderzoeken wordt weergegeven in de onderstaande tabel.
Voor de verschillende frequenties gelden de volgende definities: zeer vaak (≥ 1/10), vaak (≥ 1/100 tot
Beschrijving van geselecteerde bijwerkingen
Overgevoeligheidsreacties, waaronder gevallen van huiduitslag (inclusief urticaria), anafylactische reacties en angio-oedeem zijn gemeld.
Hypertensieve crises met encefalopathie en toevallen, waarvoor onmiddellijke medische aandacht en intensieve medische zorg nodig waren, zijn opgetreden tijdens de behandeling met epoëtine alfa, zelfs bij patiënten met eerdere normale of lage bloeddruk. Bijzondere aandacht moet worden besteed aan migraine-achtige steken als een mogelijk waarschuwingssignaal (zie rubriek 4.4).
Antilichaam-gemedieerde Pure Red Cell Aplasia is zeer zelden gemeld bij
Pediatrische patiënten met chronisch nierfalen die hemodialyse ondergaan
De blootstelling van pediatrische patiënten met chronisch nierfalen die hemodialyse ondergaan in klinische onderzoeken en postmarketingervaring is beperkt. Bij deze populatie werden geen specifieke bijwerkingen van de pediatrische populatie die niet in de bovenstaande tabel zijn vermeld, of bijwerkingen die typisch zijn voor de onderliggende ziekte, gemeld.
Melding van vermoedelijke bijwerkingen
Het melden van vermoedelijke bijwerkingen die optreden na toelating van het geneesmiddel is belangrijk omdat het een continue controle van de baten/risicoverhouding van het geneesmiddel mogelijk maakt. Beroepsbeoefenaren in de gezondheidszorg wordt verzocht alle vermoedelijke bijwerkingen te melden via het nationale meldsysteem. "adres http: //www.agenziafarmaco.gov.it/it/responsabili.
04.9 Overdosering
De therapeutische marge van epoëtine alfa is erg breed. Overdosering met epoëtine alfa kan effecten veroorzaken die in het verlengde liggen van de farmacologische effecten van het hormoon.Flebotomie kan worden uitgevoerd als te hoge hemoglobinewaarden worden gevonden.
Indien de toestand van de patiënt dit vereist, dient aanvullende ondersteunende zorg te worden verleend.
05.0 FARMACOLOGISCHE EIGENSCHAPPEN
05.1 Farmacodynamische eigenschappen
Farmacotherapeutische categorie: anti-anemisch, ATC-code: B03XA01.
Werkingsmechanisme
Erytropoëtine (EPO) is een glycoproteïnehormoon dat voornamelijk door de nieren wordt geproduceerd als reactie op hypoxie en is de belangrijkste regulator van de productie van rode bloedcellen (RBC). "EPO is betrokken bij alle fasen van erytroïde ontwikkeling en heeft zijn belangrijkste effect op het niveau van erytroïde precursors. Nadat EPO zich bindt aan zijn receptor op het celoppervlak, activeert het signaaltransductieroutes die interfereren met" apoptose en stimuleert de proliferatie van erytroïde cellen. humaan erytropoëtine (epoëtine alfa), uitgedrukt in ovariumcellen van Chinese hamsters, heeft een sequentie van 165 aminozuren die identiek is aan die van humaan urinair EPO; de 2 zijn niet te onderscheiden op basis van functionele testen Het schijnbare molecuulgewicht van erytropoëtine ligt tussen 32.000 en 40.000 daltons.
Erytropoëtine is een groeifactor die voornamelijk de aanmaak van erytrocyten stimuleert Erytropoëtinereceptoren kunnen tot expressie worden gebracht op het oppervlak van een verscheidenheid aan kankercellen.
Farmacodynamische effecten
Gezonde vrijwilligers
Na enkelvoudige doses epoëtine alfa (20.000 tot 160.000 IE subcutaan) werd een dosisafhankelijke respons waargenomen voor de onderzochte farmacodynamische markers, waaronder: reticulocyten, rode bloedcellen en hemoglobine. Er werd een gedefinieerd concentratie-temporeel profiel waargenomen met piek en terugkeer naar baseline voor veranderingen in het percentage reticulocyten. Een minder gedefinieerd profiel werd waargenomen voor erytrocyten en hemoglobine. Over het algemeen namen alle farmacodynamische markers lineair toe met doses die een maximale respons bereikten bij maximale dosisniveaus.
Aanvullende farmacodynamische studies onderzochten 40.000 IE eenmaal per week vergeleken met 150 IE/kg 3 keer per week. Ondanks de verschillen in concentratie-tijdprofielen, was de farmacodynamische respons (zoals gemeten door procentuele veranderingen in reticulocyten, hemoglobine en totaal aantal rode bloedcellen) vergelijkbaar voor deze regimes. In aanvullende onderzoeken werd het regime van 40.000 IE van epoëtine alfa eenmaal per week vergeleken met tweewekelijkse doses variërend van 80.000 tot 120.000 IE subcutaan. Op basis van de resultaten van deze farmacodynamische onderzoeken bij gezonde proefpersonen lijkt het doseringsschema van 40.000 IE eenmaal per week effectiever te zijn bij de productie van rode bloedcellen dan het tweewekelijkse schema, ondanks een overeenkomst in de productie van reticulocyten. tweewekelijkse regimes.
Chronisch nierfalen
Van epoëtine alfa is aangetoond dat het erytropoëse stimuleert bij anemische patiënten met chronisch nierfalen, waaronder dialyse- en predialysepatiënten. Het eerste bewijs van een respons op epoëtine alfa is een toename van het aantal reticulocyten binnen 10 dagen, gevolgd door een toename van het aantal rode bloedcellen, hemoglobine en hematocriet, gewoonlijk binnen 2-6 weken. De hemoglobinerespons varieert tussen patiënten en kan worden beïnvloed door ijzerafzettingen en de aanwezigheid van bijkomende medische problemen.
Door chemotherapie geïnduceerde bloedarmoede
Van epoëtine alfa, driemaal per week of eenmaal per week toegediend, is aangetoond dat het de hemoglobine verhoogt en de behoefte aan transfusies vermindert na de eerste maand van behandeling bij anemische kankerpatiënten die chemotherapie krijgen.
In een onderzoek waarin doseringsschema's van 150 IE/kg 3 maal per week en 40.000 IE eenmaal per week bij gezonde proefpersonen en bij anemische kankerpatiënten werden vergeleken, waren de temporele profielen van procentuele veranderingen in reticulocyten, hemoglobine en totaal aantal rode bloedcellen vergelijkbaar tussen de twee doseringsschema's bij gezonde en anemische kankerpatiënten. De AUC's van de respectieve farmacodynamische parameters waren vergelijkbaar tussen de doseringsschema's van 150 IE/kg 3 maal per week en 40.000 IE eenmaal per week bij zowel gezonde als anemische kankerpatiënten.
Volwassen chirurgische patiënten in een autoloog predonatieprogramma
Van epoëtine alfa is aangetoond dat het de productie van rode bloedcellen stimuleert om de autologe bloedafname te verhogen en de daling van hemoglobine te beperken bij volwassen patiënten die een grote electieve operatie moeten ondergaan en voor wie volledige perioperatieve voorbewaring niet wordt verwacht. De grootste effecten werden gezien bij patiënten met lage hemoglobinewaarden (≤ 13 g/dl).
Behandeling van volwassen patiënten die voor een grote electieve orthopedische ingreep zijn ingepland
Bij patiënten bij wie een grote electieve orthopedische operatie is ingepland met voorbehandeling van hemoglobine > 10 tot ≤ 13 g/dl, is aangetoond dat epoëtine alfa het risico op het ontvangen van allogene transfusies vermindert en het herstel van erytroïden versnelt (verhoogd hemoglobinegehalte, aantal hematocriet en reticulocyten).
Klinische werkzaamheid en veiligheid
Chronisch nierfalen
Epoëtine alfa is geëvalueerd in klinische onderzoeken bij anemische volwassen patiënten met CRF, waaronder hemodialyse- en predialysepatiënten, om anemie te behandelen en hematocriet binnen een concentratiebereik van 30 tot 36% te houden.
In klinische onderzoeken met een startdosering van 50-150 IE/kg driemaal per week, reageerde ongeveer 95% van alle patiënten met een klinisch significante verhoging van de hematocriet. Na ongeveer twee maanden therapie kregen bijna alle patiënten een transfusie. -Onafhankelijk. Eenmaal het hematocrietdoel werd bereikt, werd de onderhoudsdosis voor elke patiënt aangepast.
In de drie belangrijkste klinische onderzoeken bij volwassen dialysepatiënten was de gemiddelde onderhoudsdosis die nodig was om de hematocrietwaarde tussen 30 en 36% te houden ongeveer 75 IE/kg 3 keer per week.
In een dubbelblind, multicenter, placebogecontroleerd onderzoek naar de kwaliteit van leven van CRF-patiënten die hemodialyse ondergaan, werd een klinisch en statistisch significante verbetering aangetoond bij patiënten die met epoëtine alfa werden behandeld in vergelijking met de placebogroep, waarbij vermoeidheid, fysieke symptomen, relaties werden gemeten. en depressie (Kidney Disease Questionnaire) na zes maanden therapie. Patiënten in de epoëtine alfa-groep werden geïncludeerd in een open-label verlengingsonderzoek, waaruit bleek dat verbeteringen in hun kwaliteit van leven gedurende nog eens 12 maanden aanhielden.
Volwassen patiënten met nierinsufficiëntie die nog niet gedialyseerd worden
In klinische onderzoeken bij CRF-patiënten die niet werden gedialyseerd met epoëtine alfa, was de gemiddelde behandelingsduur bijna vijf maanden. Deze patiënten reageerden op een behandeling met epoëtine alfa op een vergelijkbare manier als bij dialysepatiënten. CRF-patiënten die geen dialyse ondergingen vertoonden dosisafhankelijkheid en een gestage toename van hematocriet wanneer epoëtine alfa zowel intraveneus als subcutaan werd toegediend. Gelijkaardige hematocrietgroeisnelheden werden waargenomen wanneer epoëtine alfa voor beide werd toegediend. Bovendien waren doses van epoëtine alfa 75 tot 150 IE/kg per week is aangetoond dat het hematocriet gedurende maximaal zes maanden op 36 tot 38% houdt.
In 2 onderzoeken met verlengde doseringsintervallen van EPREX (3 maal per week, eenmaal per week, eenmaal per twee weken en eenmaal per vier weken) behielden sommige patiënten met langere doseringsintervallen geen adequate hemoglobinewaarden en voldeden zij aan de vastgestelde ontwenningscriteria (0% eenmaal per week, 3,7% eenmaal per 2 weken en 3,3% in de groepen van eenmaal per 4 weken).
Een prospectieve, gerandomiseerde studie (CHOIR) evalueerde 1432 anemische patiënten met chronisch nierfalen die geen dialyse ondergingen. Patiënten kregen een behandeling met epoëtine alfa toegewezen met als doel een onderhoudshemoglobinegehalte van 13,5 g/dl (boven het aanbevolen hemoglobinegehalte) of 11,3 g/dl Een ernstige cardiovasculaire gebeurtenis (overlijden, myocardinfarct, beroerte of ziekenhuisopname wegens congestief hartfalen) , kwam voor bij 125 (18%) van 715 patiënten in de hogere hemoglobinegroep vergeleken met 97 (14%) bij 717 patiënten in de lagere hemoglobinegroep (risicopercentage [HR] 1,3, 95% BI: 1,0, 1,7, p = 0,03).
Cumulatieve retrospectieve analyses van klinische onderzoeken naar ESA's werden uitgevoerd bij patiënten met chronisch nierfalen (bij dialyse- en niet-dialysepatiënten, diabetici en niet-diabetici). Er werd een trend waargenomen naar een verhoogd geschat risico op mortaliteit door alle oorzaken, cardiovasculaire en cerebrovasculaire voorvallen geassocieerd met hogere cumulatieve doses ESA, ongeacht de diabetische of dialysestatus van de patiënt (zie rubriek 4.2 en rubriek 4.4).
Behandeling van patiënten met door chemotherapie geïnduceerde anemie
Epoëtine alfa is geëvalueerd in klinische onderzoeken bij volwassen anemische kankerpatiënten met lymfoïde en solide tumoren en bij patiënten die verschillende chemotherapiebehandelingen ondergaan, waaronder platina- en niet-platinabevattende behandelingen. In deze onderzoeken bleek dat epoëtine alfa, 3 keer per week en eenmaal per week toegediend, het hemoglobine verhoogt en de behoefte aan transfusies vermindert na de eerste maand van therapie bij anemische kankerpatiënten. In sommige onderzoeken werd de dubbelblinde fase gevolgd door een open-label fase waarin alle patiënten epoëtine alfa kregen en het effect werd gehandhaafd.
Beschikbare gegevens suggereren dat patiënten met hematologische maligniteiten en solide tumoren gelijkwaardig reageren op behandeling met epoëtine alfa en dat patiënten met of zonder tumorinfiltratie van het beenmerg gelijkwaardig reageren op behandeling met epoëtine alfa. De vergelijkbare intensiteit van chemotherapie in de epoëtine alfa- en placebogroepen in de chemotherapie-onderzoeken werd aangetoond door een vergelijkbaar gebied onder de neutrofieltijdcurve bij patiënten die werden behandeld met epoëtine alfa en placebo, evenals door een vergelijkbaar percentage patiënten in de behandelde groepen. met epoëtine alfa en met placebo behandelde groepen waarvan het absolute aantal neutrofielen onder de 1.000 en 500 cellen/mcL daalde
In een prospectieve, dubbelblinde, gerandomiseerde, placebo-gecontroleerde studie van 375 anemische patiënten met niet-myeloïde tumoren die niet-platina-gebaseerde chemotherapie kregen, een significante vermindering van anemie-gerelateerde symptomen (zoals asthenie, vermoeidheid en verlaagde bloeddruk) werd gevonden. activiteit), gemeten met de volgende evaluatieschalen: Functional Assessment of Cancer Therapy-Anemia (FACT-An) algemene schaal; FEIT-Een vermoeidheidsschaal en Cancer Linear Analogue Scale (CLAS).
Twee andere gerandomiseerde en placebogecontroleerde onderzoeken die bij een kleiner aantal patiënten werden uitgevoerd, lieten geen verbetering zien in de parameters voor kwaliteit van leven volgens de EORTC-QLQ-C30- en CLAS-schalen.
Overleving en tumorprogressie werden onderzocht in vijf grote gecontroleerde onderzoeken met in totaal 2.833 patiënten, waaronder vier dubbelblinde, placebogecontroleerde en één open label. In de onderzoeken werden zowel patiënten opgenomen die chemotherapie kregen (twee onderzoeken) als patiënten bij wie het gebruik van ESA's niet was geïndiceerd: anemische kankerpatiënten die geen chemotherapie kregen en patiënten met hoofd-halskanker die radiotherapie kregen Het gewenste concentratieniveau van radiotherapie hemoglobine in twee studies was> 13 g/dl, in de overige drie studies was dit 12 tot 14 g/dl In de open-label studie was er geen verschil in overleving tussen patiënten behandeld met recombinant humaan erytropoëtine en controles. In de vier gecontroleerde onderzoeken werd"risicoverhouding" voor totale overleving varieerde het van 1,25 tot 2,47 in het voordeel van controles. Deze onderzoeken toonden een onverklaarbare, statistisch significante toename van de mortaliteit aan bij kankeranemische patiënten die werden behandeld met recombinant humaan erytropoëtine in vergelijking met controles. Het algehele overlevingsresultaat van patiënten die werden behandeld met recombinant humaan erytropoëtine versus controle kon niet bevredigend worden verklaard door het verschil in de incidentie van trombose en gerelateerde complicaties.
Er werd ook een analyse op patiëntniveau uitgevoerd bij meer dan 13.900 kankerpatiënten (chemo-, radio-, radiochemio- of niet-therapie), die deelnamen aan 53 gecontroleerde klinische onderzoeken met verschillende epoëtinen. De meta-analyse van gegevens over de totale overleving gaf een stipte schatting van de hazard ratio van 1,06 in het voordeel van controles (95% BI: 1,00; 1,12; 53 onderzoeken en 13933 patiënten), terwijl voor kankerpatiënten die chemotherapie kregen, de totale overleving in termen van hazard ratio was 1,04 (95% BI: 0,97; 1,11; 38 onderzoeken en 10441 patiënten). Bovendien wijzen meta-analyses consequent op een significant verhoogd relatief risico op trombo-embolische voorvallen bij kankerpatiënten die recombinante humane erytropoëtines krijgen (zie rubriek 4.4).
Autoloog predonatieprogramma
Het effect van epoëtine alfa bij het vergemakkelijken van autologe bloeddonatie bij patiënten met een lage hematocrietwaarde (≤ 39% en zonder duidelijke anemie als gevolg van ijzertekort) die een grote orthopedische operatie moesten ondergaan, werd geëvalueerd in een dubbel placebogecontroleerd onderzoek bij 204 patiënten, en een enkele -blind placebogecontroleerd onderzoek bij 55 patiënten.
In de dubbelblinde studie werden patiënten behandeld met epoëtine alfa 600 IE/kg of placebo eenmaal daags elke 3 of 4 dagen gedurende 3 weken (totaal 6 doses). Gemiddeld konden patiënten die met epoëtine alfa werden behandeld vooraf significant meer eenheden bloed (4,5 eenheden) storten dan patiënten die met placebo (3,0 eenheden) werden behandeld.
In de enkelblinde studie werden patiënten behandeld met epoëtine alfa 300 IE/kg of 600 IE/kg of placebo eenmaal daags elke 3 of 4 dagen gedurende 3 weken (totaal 6 doses). Patiënten die met epoëtine alfa werden behandeld, waren ook in staat om vooraf significante extra eenheden bloed te storten (epoëtine alfa 300 IE/kg = 4,4 eenheden; epoëtine alfa 600 IE/kg = 4,7 eenheden) in vergelijking met patiënten die werden behandeld met placebo (2,9 eenheden).
Behandeling met epoëtine alfa verminderde het risico op blootstelling aan allogene bloed met 50% in vergelijking met patiënten die niet met epoëtine alfa werden behandeld.
Grote electieve orthopedische chirurgie
Het effect van epoëtine alfa (300 IE/kg of 100 IE/kg) op blootstelling aan allogene bloedtransfusies werd geëvalueerd in een dubbelblind, placebogecontroleerd klinisch onderzoek bij volwassen patiënten met ijzertekort die wachtten op een operatie. of knieoperatie Epoëtine alfa werd subcutaan toegediend gedurende 10 dagen voorafgaand aan de operatie, op de dag van de operatie en gedurende vier dagen na de operatie. Patiënten werden geclassificeerd op basis van hun baseline hemoglobine (≤ 10 g / dL,> 10 tot ≤ 13 g / dL en> 13 g / dL).
Epoëtine alfa 300 IE/kg verminderde significant het risico op allogene transfusies bij patiënten met hemoglobine vóór de behandeling van > 10 tot ≤ 13 g/dL. 16% van de patiënten behandeld met epoëtine alfa 300 IE/kg, 23% met epoëtine alfa 100 IE/kg en 45% met placebo hadden transfusie nodig.
Een open-label studie met parallelle groepen bij volwassen proefpersonen met ijzertekort met een hemoglobinegehalte van 10 tot ≤ 13 g/dl voorafgaand aan de behandeling die wachtten op een orthopedische heup- of knieoperatie, vergeleek behandeling met epoëtine alfa 300 IE/kg dagelijks subcutaan gedurende 10 dagen vóór de operatie, op de dag van de operatie en gedurende vier dagen na de operatie, met behandeling met epoëtine alfa 600 IE/kg subcutaan eenmaal per week gedurende 3 weken voorafgaand aan de operatie en op de dag van de operatie.
Van voorbehandeling tot pre-operatie, was de gemiddelde toename van hemoglobine in de wekelijkse groep van 600 IE/kg (1,44 g/dl) het dubbele van die waargenomen in de groep van 300 IE/kg per dag (0,73 g/dl). De gemiddelde hemoglobinespiegels waren vergelijkbaar in de twee behandelingsgroepen gedurende de postoperatieve periode.
De erytropoëtische respons waargenomen in beide behandelingsgroepen leidde tot vergelijkbare transfusiesnelheden (16% in de 600 IE/kg/week-groep en 20% in de 300 IE/kg/dag-groep)
Pediatrische populatie
Chronisch nierfalen
Epoëtine alfa werd geëvalueerd in een 52 weken durend open-label, niet-gerandomiseerd, open dosisbereik bij pediatrische CRF-patiënten die hemodialyse ondergingen. De gemiddelde leeftijd van de patiënten die deelnamen aan het onderzoek was 11,6 jaar (bereik 0,5 tot - 20,1 jaar).
Epoëtine alfa werd intraveneus toegediend met 75 IE/kg/week in 2 of 3 verdeelde doses na dialyse, getitreerd van 75 IE/kg/week met intervallen van 4 weken (tot een maximum van 300 IE/kg/week.), om een hemoglobinestijging van 1 g / dl / maand te bereiken Het gewenste hemoglobinegehalte was 9,6 tot 11,2 g / dl Eenentachtig procent van de patiënten bereikte het beoogde hemoglobinegehalte. Mediane tijd tot bestemming was 11 weken, mediane dosis tot bestemming was 150 IE/kg/week. Van de patiënten die het doel bereikten, deed 90% dat met een doseringsschema van 3 keer per week.
Na 52 weken bleef 57% van de patiënten in het onderzoek en kreeg een gemiddelde dosis van 200 IE/kg/week.
05.2 Farmacokinetische eigenschappen
Absorptie
Na subcutane toediening pieken de serumspiegels van epoëtine alfa tussen 12 en 18 uur na toediening. Er was geen accumulatie na toediening van meerdere wekelijkse doses van 600 IE/kg subcutaan toegediend.
De absolute biologische beschikbaarheid van subcutaan geïnjecteerde epoëtine alfa is ongeveer 20% bij gezonde proefpersonen.
Verdeling
Het gemiddelde distributievolume is 49,3 ml/kg na intraveneuze toediening van 50 en 100 IE/kg bij gezonde proefpersonen. Na intraveneuze toediening van epoëtine alfa bij personen met chronisch nierfalen varieerde het distributievolume van 57-107 ml/kg na enkelvoudige toediening (12 IE/kg) tot 42-64 ml/kg na toediening van meerdere doses (48-192 IE /kg) respectievelijk. Dientengevolge is het distributievolume iets groter dan het plasmavolume.
Eliminatie
De halfwaardetijd van epoëtine alfa na intraveneuze toediening van meerdere doses is bij gezonde proefpersonen ongeveer 4 uur. De halfwaardetijd na subcutane toediening wordt geschat op ongeveer 24 uur bij gezonde proefpersonen.
De gemiddelde CL/F-waarde voor de regimes van 150 IE/kg 3 maal per week en 40.000 IE eenmaal per week bij gezonde proefpersonen waren respectievelijk 31,2 en 12,6 ml/u/kg. De gemiddelde CL/F-waarde voor de regimes van 150 IE/kg 3 maal per week en 40.000 IE eenmaal per week bij anemische kankerpatiënten was respectievelijk 45,8 en 11,3 ml/u/kg. Bij de meerderheid van de anemische kankerpatiënten die cyclische chemotherapie kregen, was de CL/F lager na subcutane doses van 40.000 IE eenmaal per week en 150 IE/kg 3 keer per week, vergeleken met de waarden voor gezonde proefpersonen.
Lineariteit / niet-lineariteit
Bij gezonde proefpersonen werd een dosisproportionele verhoging van de serumconcentraties van epoëtine alfa waargenomen na intraveneuze toediening van 150 en 300 IE/kg, 3 keer per week. Enkelvoudige subcutane doses van 300-2400 IE/kg epoëtine alfa resulteerden in een lineair verband tussen gemiddelde Cmax en dosis en tussen gemiddelde AUC en dosis. Er werd een omgekeerd verband waargenomen tussen de opruimingzichtbaar en gedoseerd bij gezonde proefpersonen.
In onderzoeken naar de verlenging van het interval tussen doses (40.000 IE eenmaal per week en 80.000, 100.000 en 120.000 IE tweewekelijks), werd een lineair maar niet-dosisproportioneel verband waargenomen tussen gemiddelde Cmax en dosis el "Gemiddelde AUC en dosis bij steady-state .
Farmacokinetische / farmacodynamische relatie
Epoëtine alfa vertoont een dosisgerelateerd effect op hematologische parameters dat onafhankelijk is van de toedieningsweg.
Pediatrische populatie
Een halfwaardetijd van ongeveer 6,2-8,7 uur na meervoudige intraveneuze toediening van epoëtine alfa is gemeld bij pediatrische patiënten met chronisch nierfalen.Het farmacokinetische profiel van epoëtine alfa bij kinderen en adolescenten lijkt vergelijkbaar te zijn met dat bij volwassenen.
Nierfalen
Bij patiënten met chronisch nierfalen is de halfwaardetijd van epoëtine alfa, intraveneus toegediend, iets langer, ongeveer 5 uur, in vergelijking met gezonde proefpersonen.
05.3 Gegevens uit het preklinisch veiligheidsonderzoek
In toxicologische onderzoeken met herhaalde dosering bij honden en ratten, maar niet bij apen, werd behandeling met epoëtine alfa in verband gebracht met een subklinische aandoening van beenmergfibrose. Beenmergfibrose is een bekende complicatie van chronisch nierfalen bij mensen en kan verband houden met secundaire hyperparathyreoïdie of wordt veroorzaakt door onbekende factoren. In een onderzoek bij hemodialysepatiënten die gedurende 3 jaar met epoëtine alfa werden behandeld, was de incidentie van beenmergfibrose niet hoger dan in de groep controledialysepatiënten die niet met epoëtine alfa werden behandeld.
Epoëtine alfa induceert geen genmutaties in bacteriën (Ames-test), chromosomale aberratie in zoogdiercellen, micronuclei bij muizen of genmutaties op de HGPRT-locus.
Er zijn geen carcinogeniteitsonderzoeken op lange termijn uitgevoerd. Tegenstrijdige resultaten in de literatuur op basis van de gegevens in vitro uit humane tumormonsters, suggereren dat erytropoëtines een rol kunnen spelen bij tumorproliferatie.Dit is van onzekere betekenis in de klinische praktijk.
In celculturen van menselijke beenmergcellen stimuleert epoëtine alfa specifiek erytropoëse en is er geen sprake van leukopoëse.Er is geen cytotoxische activiteit van epoëtine alfa waargenomen op beenmergcellen.
In dierstudies is aangetoond dat epoëtine alfa het foetale lichaamsgewicht verlaagt, de ossificatie vertraagt en de foetale mortaliteit verhoogt wanneer het wordt toegediend in wekelijkse doses van ongeveer 20 maal de aanbevolen wekelijkse dosis bij mensen. Deze veranderingen worden beschouwd als secundair aan de afname van het lichaamsgewicht van de moeder en de significantie voor mensen bij therapeutische doses is niet bekend.
06.0 FARMACEUTISCHE INFORMATIE
06.1 Hulpstoffen
Polysorbaat 80
Glycine
Water voor injecties.
Natriummonobasisch fosfaatdihydraat
Dibasisch natriumfosfaatdihydraat
Natriumchloride
06.2 Incompatibiliteit
Bij gebrek aan onderzoek naar onverenigbaarheden, mag het geneesmiddel niet met andere producten worden gemengd.
06.3 Geldigheidsduur
18 maanden.
06.4 Speciale voorzorgsmaatregelen bij bewaren
Bewaren in de koelkast (2 ° C - 8 ° C). Dit temperatuurbereik moet worden gegarandeerd tot het moment van toediening aan de patiënt.
Bewaren in de originele verpakking om het product tegen licht te beschermen.
Niet invriezen of schudden.
Voor poliklinisch gebruik kan het geneesmiddel uit de koelkast worden gehaald, zonder te worden vervangen, gedurende maximaal 3 dagen bij een temperatuur van maximaal 25°C. Als het geneesmiddel aan het einde van deze periode niet is gebruikt, moet het worden weggegooid.
06.5 Aard van de primaire verpakking en inhoud van de verpakking
0,3 ml (3.000 IE) oplossing voor injectie in een voorgevulde spuit (type I-glas) met zuiger (rubber met tefloncoating) en naald met behuizing (rubber met polypropyleencoating) en PROTECS-naaldbeveiliging (polycarbonaat) bevestigd aan de spuit - pak van 1.
0,4 ml (4000 IE) in een voorgevulde spuit (type I glas) met zuiger (rubber met tefloncoating) en naald met behuizing (rubber met polypropyleen coating) en PROTECS naaldbeveiliging (polycarbonaat) bevestigd aan de spuit - verpakking van 1.
0,5 ml (5.000 IE) oplossing voor injectie in een voorgevulde spuit (type I-glas) met zuiger (rubber met tefloncoating) en naald met behuizing (rubber met polypropyleencoating) en PROTECS-naaldbeveiliging (polycarbonaat) bevestigd aan de spuit - pak van 1.
0,6 ml (6.000 IE) oplossing voor injectie in een voorgevulde spuit (type I glas) met zuiger (rubber met tefloncoating) en naald met behuizing (rubber met polypropyleen coating) en PROTECS naaldbeveiliging (polycarbonaat) bevestigd aan de spuit - pak van 1.
0,8 ml (7.000 IE) oplossing voor injectie in een voorgevulde spuit (type I-glas) met zuiger (rubber met tefloncoating) en naald met behuizing (rubber met polypropyleencoating) en PROTECS-naaldbeveiliging (polycarbonaat) bevestigd aan de spuit - pak van 1.
1,0 ml (10.000 IE) oplossing voor injectie in een voorgevulde spuit (type I-glas) met zuiger (rubber met tefloncoating) en naald met behuizing (rubber met polypropyleencoating) en PROTECS-naaldbeveiliging (polycarbonaat) bevestigd aan de spuit - pak van 1.
06.6 Instructies voor gebruik en verwerking
Het product mag niet worden gebruikt en moet worden weggegooid:
• Als de verzegeling verbroken is;
• Als de oplossing gekleurd is of in aanwezigheid van deeltjes;
• Als bevriezing is opgetreden of wordt vermoed;
• Als er een koelkaststoring is.
Wegwerpproduct. Dien niet meer dan één dosis per spuit toe nadat de ongewenste hoeveelheid oplossing uit de spuit is verwijderd. Zie rubriek 3. Hoe gebruikt u EPREX (injectie-instructies) van de bijsluiter.
De voorgevulde spuiten zijn uitgerust met een PROTECS-naaldbeveiliging om het risico van naaldprikken te voorkomen.De bijsluiter bevat alle informatie voor het gebruik en de omgang met de voorgevulde spuiten met het veiligheidsapparaat.PROTECS-naald.
Ongebruikte medicijnen en afval afkomstig van dit medicijn moeten worden weggegooid in overeenstemming met de lokale regelgeving.
07.0 HOUDER VAN DE VERGUNNING VOOR HET IN DE HANDEL BRENGEN
Janssen-Cilag SpA, via M.Buonarroti, 23 - 20093 Cologno Monzese (MI)
08.0 NUMMER VAN DE VERGUNNING VOOR HET IN DE HANDEL BRENGEN
027015167 - "10.000 IE/ML oplossing voor injectie in voorgevulde spuit" 1 spuit van 3.000 IE/0.3ML
027015179 - "10.000 IE/ML oplossing voor injectie in voorgevulde spuit" 1 spuit van 4.000 IE/0,4ML
027015231 - "10.000 IE/ML oplossing voor injectie in voorgevulde spuit" 1 spuit van 5.000 IE/0,5ML
027015243 - "10.000 IE/ML oplossing voor injectie in voorgevulde spuit" 1 spuit van 6.000 IE/0.6ML
027015268 - "10.000 IE/ML oplossing voor injectie in voorgevulde spuit" 1 spuit van 8.000 IE/0,8ML
027015181 - "10.000 IE / ML oplossing voor injectie in voorgevulde spuit" 1 spuit van 10.000 IE / 1ML
09.0 DATUM VAN EERSTE VERGUNNING OF VERLENGING VAN DE VERGUNNING
mei 1989
AIC-verlenging: 4 augustus 2008
10.0 DATUM VAN HERZIENING VAN DE TEKST
08/2015