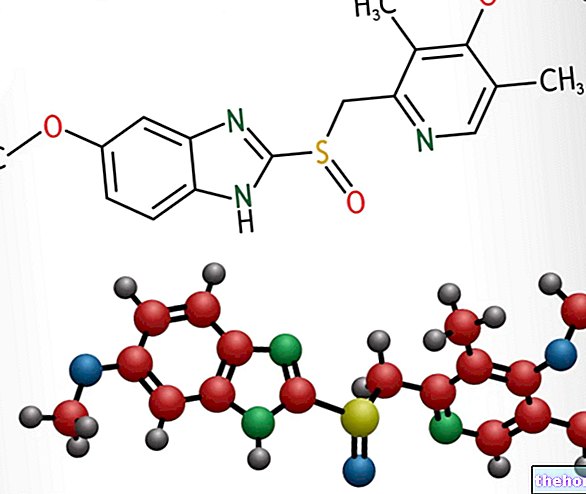
Actieve ingrediënten: Levetiracetam
Matever 100 mg/ml concentraat voor oplossing voor infusie
Matever bijsluiters zijn beschikbaar voor verpakkingsgrootten:- Matever 250 mg filmomhulde tabletten, Matever 500 mg filmomhulde tabletten, Matever 750 mg filmomhulde tabletten, Matever 1000 mg filmomhulde tabletten
- Matever 100 mg/ml concentraat voor oplossing voor infusie
Indicaties Waarom wordt Matever gebruikt? Waar is het voor?
Levetiracetam-concentraat is een anti-epilepticum (een geneesmiddel dat wordt gebruikt om aanvallen te behandelen).
Matever wordt gebruikt:
- als monotherapie bij volwassenen en adolescenten vanaf 16 jaar met nieuw gediagnosticeerde epilepsie, om een bepaalde vorm van epilepsie te behandelen. Epilepsie is een aandoening waarbij de patiënt herhaalde aanvallen (aanvallen) heeft.Leviteracetam wordt gebruikt voor de vorm van epilepsie waarbij de aanvallen aanvankelijk een deel van de hersenen aantasten, maar zich later kunnen uitbreiden naar een groter gebied aan weerszijden van de hersenen ( partieel beginnende aanvallen met of zonder secundaire generalisatie). Leviteracetam is door uw arts aan u gegeven om het aantal aanvallen te verminderen
- als aanvulling op andere anti-epileptica voor de behandeling van: - partieel beginnende aanvallen, met of zonder generalisatie, bij volwassenen, adolescenten en kinderen vanaf 4 jaar - myoclonische aanvallen (korte, schokkerige schokken van een spier of spiergroep) bij volwassenen en adolescenten vanaf 12 jaar met juveniele myoclonische epilepsie - primaire gegeneraliseerde tonisch-clonische aanvallen (ernstige aanvallen waaronder bewustzijnsverlies) bij volwassenen en adolescenten en kinderen vanaf 12 jaar met idiopathische gegeneraliseerde epilepsie (het type epilepsie waarvan wordt aangenomen dat het het gevolg is van genetische oorzaken).
Matever concentraat is een alternatief voor patiënten wanneer toediening van het orale anti-epilepticum Matever tijdelijk niet mogelijk is.
Contra-indicaties Wanneer Matever niet mag worden gebruikt
Je gebruikt geen Matever
- Als u allergisch bent voor levetiracetam, pyrrovidonderivaten of voor één van de andere bestanddelen van dit geneesmiddel.
Voorzorgen bij gebruik Wat u moet weten voordat u Matever inneemt
Praat met uw arts voordat u Matever gebruikt
- Als u nierproblemen heeft, volg dan de instructies van uw arts op. Deze laatste kan beslissen of de dosis moet worden aangepast.
- Als u een vertraging in de groei of een onverwachte ontwikkeling van de puberteit bij uw kind opmerkt, neem dan contact op met uw arts.
- Een beperkt aantal mensen dat werd behandeld met anti-epileptica zoals Matever, heeft gedachten gehad over schade of zelfmoord. Als u symptomen van depressie en/of zelfmoordgedachten heeft, neem dan contact op met uw arts.
Kinderen en adolescenten
Matever is niet geïndiceerd bij kinderen of adolescenten jonger dan 16 jaar als enige behandeling (monotherapie).
Interacties Welke medicijnen of voedingsmiddelen kunnen het effect van Matever veranderen
Vertel het uw arts of apotheker als u andere geneesmiddelen gebruikt of kort geleden heeft gebruikt.
Gebruik geen macrogol (of geneesmiddelen die als laxeermiddel worden gebruikt) één uur vóór en één uur na inname van levetiracetam, omdat dit minder effectief kan zijn.
Waarschuwingen Het is belangrijk om te weten dat:
Zwangerschap en borstvoeding
Als u zwanger bent of borstvoeding geeft, denkt zwanger te zijn of zwanger wilt worden, vraag dan uw arts of apotheker om advies voordat u dit geneesmiddel inneemt.
Matever mag niet tijdens de zwangerschap worden gebruikt, tenzij strikt noodzakelijk. Een risico op aangeboren afwijkingen voor de foetus kan niet volledig worden uitgesloten. Matever heeft ongewenste reproductieve effecten aangetoond in dierstudies met hogere doseringen dan nodig zijn om aanvallen onder controle te houden.
Borstvoeding wordt tijdens de behandeling niet aanbevolen.
Rijvaardigheid en het gebruik van machines
Matever kan de rijvaardigheid of het vermogen om gereedschap of machines te gebruiken verminderen, aangezien het slaperigheid kan veroorzaken. Dit is waarschijnlijker aan het begin van de behandeling of na een dosisverhoging.
U mag niet autorijden of machines bedienen totdat u heeft geverifieerd dat uw vermogen om deze activiteiten uit te voeren niet wordt beïnvloed.
Matever bevat natrium
Een enkele maximale dosis Matever-concentraat bevat 2,49 mmol (of 57,21 mg) natrium (0,8 mmol (of 19 mg) natrium per injectieflacon). Hiermee moet rekening worden gehouden bij patiënten die een natriumarm dieet volgen.
Dosering en wijze van gebruik Hoe Matever te gebruiken: Dosering
Een arts of verpleegkundige zal u Matever toedienen als een intraveneuze infusie.
De intraveneuze formulering is een alternatief voor orale toediening. U kunt direct zonder dosisaanpassing overschakelen van filmomhulde tabletten of drank naar intraveneuze formulering of vice versa. De totale dagelijkse dosis en de toedieningsfrequentie blijven hetzelfde.
Matever moet tweemaal per dag worden toegediend, eenmaal 's ochtends en eenmaal 's avonds, elke dag op ongeveer hetzelfde tijdstip.
Monotherapie
Dosis bij volwassenen en adolescenten (vanaf 16 jaar)
Typische dosis: tussen 1000 mg en 3000 mg per dag.
Wanneer u voor het eerst Matever gaat gebruiken, zal uw arts u gedurende 2 weken een lagere dosis voorschrijven voordat u de gebruikelijke lagere dosis krijgt.
Aanvullende therapie
Dosis bij volwassenen en adolescenten (12 tot 17 jaar) met een gewicht van 50 kg of meer
Typische dosis: tussen 1000 mg en 3000 mg per dag.
Dosis voor kinderen (4 tot 11 jaar) en adolescenten (12 tot 17 jaar) met een gewicht van minder dan 50 kg
Typische dosering: tussen 20 mg per kg lichaamsgewicht en 60 mg per kg lichaamsgewicht per dag.
Wijze van toediening en wijze van toediening
Matever is voor intraveneus gebruik. De aanbevolen dosis moet worden verdund in ten minste 100 ml van een compatibel verdunningsmiddel en gedurende 15 minuten worden toegediend.
Duur van de behandeling
Er is geen ervaring met intraveneuze toediening van levetiracetam langer dan 4 dagen.
Als u stopt met het innemen van Matever
Als de behandeling wordt stopgezet, moet Matever geleidelijk worden stopgezet om meer aanvallen te voorkomen.
Als uw arts besluit uw behandeling met Matever stop te zetten, zal hij u instrueren om geleidelijk te stoppen.
Als u nog vragen heeft over het gebruik van dit geneesmiddel, neem dan contact op met uw arts of verpleegkundige.
Bijwerkingen Wat zijn de bijwerkingen van Matever
Zoals alle geneesmiddelen kan ook dit geneesmiddel bijwerkingen hebben, al krijgt niet iedereen daarmee te maken.
De meest frequent gemelde bijwerkingen waren nasofaryngitis, slaperigheid, hoofdpijn, vermoeidheid en duizeligheid. Aan het begin van de behandeling of bij een dosisverhoging.
Bijwerkingen zoals slaperigheid, vermoeidheid en duizeligheid kunnen vaker voorkomen. Deze effecten zouden echter na verloop van tijd moeten afnemen.
Zeer vaak: kan voorkomen bij meer dan 1 op de 10 mensen
- nasofaryngitis;
- slaperigheid, hoofdpijn;
Vaak: kan voorkomen bij 1 tot 10 patiënten op 100 mensen
- anorexia (verlies van eetlust);
- depressie, vijandigheid of agressie, angst, slapeloosheid, nervositeit of prikkelbaarheid;
- convulsies, evenwichtsstoornissen, duizeligheid (gevoel van onvastheid), lethargie (gebrek aan energie en enthousiasme), tremor (onwillekeurige tremoren);
- duizeligheid (gevoel van rotatie);
- hoest;
- buikpijn, diarree, dyspepsie (indigestie), braken, misselijkheid;
- uitslag;
- asthenie / vermoeidheid (zwak voelen).
Soms: kan voorkomen bij 1 tot 10 patiënten op 1000 mensen
- afname van het aantal bloedplaatjes in het bloed, afname van het aantal witte bloedcellen;
- gewichtsverlies, gewichtstoename;
- zelfmoordpoging en zelfmoordgedachten, psychische stoornis, abnormaal gedrag, hallucinaties, woede, verwarring, paniekaanval, emotionele labiliteit/stemmingswisselingen, opwinding;
- geheugenverlies (geheugenverlies), geheugenstoornis (vergeetachtigheid), abnormale coördinatie / ataxie (verminderde motorische coördinatie), paresthesie (tintelingen), verminderde aandacht (concentratieverlies);
- diplopie (dubbelzien), wazig zien;
- verhoogde/abnormale waarden bij leverfunctietesten,
- haaruitval, eczeem, jeuk;
- spierzwakte, myalgie (spierpijn);
- trauma;
Zelden: kan voorkomen bij 1 tot 10 patiënten op 10.000 mensen
- infectie;
- afname van het aantal van alle soorten bloedcellen;
- ernstige allergische reacties (DRESS, anafylactische reactie (ernstige en belangrijke allergische reactie), Quincke-oedeem (zwelling van het gezicht, de lippen, de tong en de keel);
- afname van de natriumconcentratie in het bloed;
- zelfmoord, persoonlijkheidsstoornis (gedragsproblemen), veranderd denken (langzaam denken, onvermogen om zich te concentreren);
- oncontroleerbare spierspasmen van het hoofd, de romp en de ledematen, moeite met het beheersen van bewegingen, hyperkinese (hyperactiviteit);
- pancreatitis;
- leverfalen, hepatitis;
- huiduitslag die blaren kan vormen en eruitziet als kleine doelwitten (centrale donkere vlek omgeven door een "lichter gebied, met een donkere ring rond de rand) (erythema multiforme), een wijdverspreide uitslag met blaren en vervellen van de huid, vooral rond de mond, neus, ogen en geslachtsdelen (Stevens-Johnson-syndroom) en een ernstigere vorm die huidschilfers veroorzaakt op meer dan 30% van het lichaamsoppervlak (toxische epidermale necrolyse).
Melding van bijwerkingen
Krijgt u last van bijwerkingen, neem dan contact op met uw arts of apotheker.Dit geldt ook voor mogelijke bijwerkingen die niet in deze bijsluiter staan. U kunt bijwerkingen ook rechtstreeks melden via het nationale meldsysteem op www.agenziafarmaco.it/it/responsabili Door bijwerkingen te melden, kunt u ons helpen meer informatie te verkrijgen over de veiligheid van dit geneesmiddel.
Vervaldatum en retentie
Buiten het zicht en bereik van kinderen houden.
Gebruik dit geneesmiddel niet meer na de vervaldatum die staat vermeld op de injectieflacon en de doos na EXP: De vervaldatum verwijst naar de laatste dag van die maand.
Voor dit geneesmiddel zijn er geen speciale bewaarcondities.
Gooi geneesmiddelen niet weg via het afvalwater of met huishoudelijk afval. Vraag uw apotheker wat u met geneesmiddelen moet doen die u niet meer gebruikt. Dit helpt het milieu te beschermen.
Andere informatie
Wat bevat Matever
Het actieve ingrediënt wordt levetiracetam genoemd. Elke ml oplossing voor infusie bevat 100 mg levetiracetam.
De andere componenten zijn: natriumacetaat, ijsazijn, natriumchloride, water voor injecties.
Beschrijving van hoe Matever eruit ziet en inhoud van het pakket
Matever concentraat voor oplossing voor infusie (Matever concentraat) is een heldere, kleurloze en steriele vloeistof.
Matever concentraat 5 ml injectieflacon is verpakt in een kartonnen doos met 10 injectieflacons.
De volgende informatie is alleen bedoeld voor beroepsbeoefenaren in de gezondheidszorg:
Instructies voor het juiste gebruik van Matever vindt u in paragraaf 3.
Eén injectieflacon Matever-concentraat bevat 500 mg levetiracetam (5 ml van 100 mg/ml concentraat).
Zie Tabel 1 voor de bereiding en aanbevolen toediening van Matever-concentraat om een totale dagelijkse dosis van 500 mg, 1000 mg, 2000 mg of 3000 mg te bereiken, verdeeld over twee doses.
Tabel 1. Bereiding en toediening van Matever-concentraat.
Dit geneesmiddel is uitsluitend bedoeld voor eenmalig gebruik en alle ongebruikte oplossing moet worden weggegooid.
Houdbaarheid tijdens gebruik: Vanuit microbiologisch oogpunt moet het product onmiddellijk na verdunning worden gebruikt. Indien niet onmiddellijk gebruikt, zijn de bewaartijden en -condities voorafgaand aan gebruik de verantwoordelijkheid van de gebruiker en mogen deze normaal gesproken niet langer zijn dan 24 uur bij een temperatuur tussen 2 en 8 ° C, tenzij de verdunning is uitgevoerd onder gecontroleerde en gevalideerde aseptische omstandigheden.
Matever-concentraat was fysiek compatibel en chemisch stabiel gedurende ten minste 24 uur wanneer het werd gemengd met de volgende verdunningsmiddelen en bewaard in PVC-zakken bij een gecontroleerde kamertemperatuur van 15-25 ° C.
Verdunners:
- Natriumchloride (0,9%) voor injectie
- Ringer's lactaatinjectie
- Dextrose 5% injecteerbaar preparaat.
Bron Bijsluiter: AIFA (Italiaans Geneesmiddelenbureau). Inhoud gepubliceerd in januari 2016. De aanwezige informatie is mogelijk niet up-to-date.
Om toegang te hebben tot de meest actuele versie, is het raadzaam om de website van AIFA (Italian Medicines Agency) te bezoeken. Disclaimer en nuttige informatie.
01.0 NAAM VAN HET GENEESMIDDEL
MATEVER 100 MG / ML CONCENTRAAT VOOR OPLOSSING VOOR INFUSIE
02.0 KWALITATIEVE EN KWANTITATIEVE SAMENSTELLING
Elke ml bevat 100 mg levetiracetam.
Elke injectieflacon van 5 ml bevat 500 mg levetiracetam.
Hulpstoffen met bekende effecten:
Elke ml bevat 3,81 mg natrium.
Voor de volledige lijst van hulpstoffen, zie rubriek 6.1.
03.0 FARMACEUTISCHE VORM
Concentraat voor oplossing voor infusie (steriel concentraat).
Helder, kleurloos concentraat.
04.0 KLINISCHE INFORMATIE
04.1 Therapeutische indicaties
Matever is geïndiceerd als monotherapie voor de behandeling van partieel beginnende aanvallen met of zonder secundaire generalisatie bij volwassenen en adolescenten vanaf 16 jaar met nieuw gediagnosticeerde epilepsie.
Matever is geïndiceerd als aanvullende therapie
• bij de behandeling van partieel beginnende aanvallen met of zonder secundaire generalisatie bij adolescente volwassenen en kinderen vanaf 4 jaar met epilepsie
• bij de behandeling van myoclonische aanvallen bij volwassenen en adolescenten vanaf 12 jaar met juveniele myoclonische epilepsie
• bij de behandeling van primaire gegeneraliseerde tonisch-clonische aanvallen bij volwassenen en adolescenten vanaf 12 jaar met idiopathische gegeneraliseerde epilepsie.
Matever concentraat is een alternatief voor patiënten wanneer orale toediening tijdelijk niet mogelijk is.
04.2 Dosering en wijze van toediening
Dosering
Monotherapie voor volwassenen en adolescenten vanaf 16 jaar
De aanbevolen startdosering is 250 mg tweemaal daags, die na twee weken moet worden verhoogd tot een therapeutische initiële dosis van 500 mg tweemaal daags. Op basis van de klinische respons kan de dosis verder worden verhoogd met 250 mg tweemaal daags om de twee weken. De maximale dosering is tweemaal daags 1500 mg.
Aanvullende therapie voor volwassenen (≥ 18 jaar) en adolescenten (12 tot 17 jaar) die 50 kg of meer wegen
De therapeutische aanvangsdosis is tweemaal daags 500 mg. Deze dosis kan worden gestart op de eerste dag van de behandeling.
Op basis van de klinische respons en verdraagbaarheid kan de dagelijkse dosis worden verhoogd tot maximaal 1500 mg tweemaal daags. Doseringsaanpassingen kunnen elke twee tot vier weken worden gedaan in verhogingen of verlagingen van 500 mg tweemaal daags.
Duur van de behandeling
Er is geen ervaring met intraveneuze toediening van levetiracetam langer dan 4 dagen.
Speciale populaties
Ouderen (65 jaar en ouder)
Aanpassing van de dosering wordt aanbevolen bij oudere patiënten met een verminderde nierfunctie (zie "Nierfunctiestoornis" hieronder).
Nierfunctiestoornis
De dagelijkse dosis moet worden aangepast aan de nierfunctie.
Raadpleeg voor volwassen patiënten de volgende tabel en pas de dosering aan zoals aangegeven. Om deze doseringstabel te gebruiken is het noodzakelijk om de creatinineklaring (CLcr) van de patiënt in ml/min te schatten. CLcr in ml/min kan worden berekend uit de bepaling van serumcreatinine (mg/dl) met behulp van, voor volwassenen en adolescenten die 50 kg of meer wegen, de volgende formule:
Bovendien wordt CLcr als volgt aangepast voor het lichaamsoppervlak (BSA):
Dosisaanpassing voor volwassen en adolescente patiënten die meer dan 50 kg wegen met een verminderde nierfunctie:
Een oplaaddosis van 750 mg wordt aanbevolen op de eerste dag van de behandeling met levetiracetam.
Na dialyse wordt een aanvullende dosis tussen 250 en 500 mg aanbevolen.
Voor kinderen met een verminderde nierfunctie moet de dosis levetiracetam worden aangepast op basis van de nierfunctie, aangezien de klaring van levetiracetam verband houdt met de nierfunctie. Deze aanbeveling is gebaseerd op een onderzoek dat is uitgevoerd bij volwassen patiënten met een verminderde nierfunctie.
Bij jonge adolescenten en kinderen kan de CLcr, in ml/min/1,73 m2, worden geschat aan de hand van de bepaling van serumcreatinine (in mg/dL) met behulp van de volgende formule (formule van Schwartz):
ks = 0,55 bij kinderen jonger dan 13 jaar en bij adolescente vrouwen; ks = 0,7 bij adolescente mannen.
Dosisaanpassing voor kinderen en adolescenten die minder dan 50 kg wegen met een verminderde nierfunctie:
Een oplaaddosis van 15 mg/kg (0,15 ml/kg) wordt aanbevolen op de eerste dag van de behandeling met levetiracetam.
Na dialyse wordt een aanvullende dosis van 5 tot 10 mg/kg (0,05 tot 0,10 ml/kg) aanbevolen.
leverfunctiestoornis
Bij patiënten met een lichte tot matige leverfunctiestoornis is geen dosisaanpassing nodig. Bij patiënten met een ernstige leverfunctiestoornis kan de creatinineklaring de mate van nierinsufficiëntie onderschatten. Daarom wordt een verlaging van de dagelijkse onderhoudsdosis met 50% aanbevolen wanneer de creatinineklaring 2 is.
Pediatrische populatie
Uw arts moet de meest geschikte farmaceutische vorm en sterkte voorschrijven op basis van uw leeftijd, gewicht en dosis.
Monotherapie
De veiligheid en werkzaamheid van Matever, gegeven als monotherapie aan kinderen en adolescenten jonger dan 16 jaar, zijn niet vastgesteld.
Er zijn geen gegevens beschikbaar.
Add-on therapie voor kinderen van 4 tot 11 jaar en adolescenten (12 tot 17 jaar) met een gewicht van minder dan 50 kg
De therapeutische aanvangsdosis is tweemaal daags 10 mg/kg.
Op basis van de klinische respons en verdraagbaarheid kan de dosis worden verhoogd tot tweemaal daags 30 mg/kg. Doseringsaanpassingen mogen de verhogingen of verlagingen van 10 mg/kg tweemaal daags om de twee weken niet overschrijden. De laagste effectieve dosis moet worden gebruikt.
De dosis bij kinderen die 50 kg of meer wegen is dezelfde als bij volwassenen.
Aanbevolen dosering voor kinderen en adolescenten:
Kinderen die 25 kg of minder wegen, dienen bij voorkeur de behandeling te starten met Matever 100 mg/ml drank.
De dosis bij kinderen en adolescenten die 50 kg of meer wegen, is dezelfde als bij volwassenen.
Add-on therapie voor zuigelingen en kinderen jonger dan 4 jaar
De veiligheid en werkzaamheid van Matever concentraat voor oplossing voor infusie bij zuigelingen en kinderen jonger dan 4 jaar zijn niet vastgesteld.
De momenteel beschikbare gegevens worden beschreven in rubrieken 4.8, 5.1 en 5.2, maar er kan geen doseringsadvies worden gegeven.
Wijze van toediening
Matever-therapie kan worden gestart met intraveneuze of orale toediening.
Conversie naar intraveneus van orale toediening of vice versa kan direct zonder titratie worden gedaan. De totale dagelijkse dosis en frequentie van toediening moeten worden gehandhaafd.
Matever-concentraat is alleen voor intraveneus gebruik en de aanbevolen dosis moet worden verdund in ten minste 100 ml van een verenigbaar verdunningsmiddel en intraveneus worden toegediend als een 15 minuten durende intraveneuze infusie (zie rubriek 6.6).
04.3 Contra-indicaties
Overgevoeligheid voor de werkzame stof of voor andere pyrrolidonderivaten of voor één van de in rubriek 6.1 vermelde hulpstoffen.
04.4 Bijzondere waarschuwingen en passende voorzorgen bij gebruik
Stopzetting van de behandeling
In overeenstemming met de huidige klinische praktijk wordt een geleidelijke stopzetting aanbevolen als de behandeling met Matever moet worden stopgezet (bijv. bij volwassenen en adolescenten die meer dan 50 kg wegen: afname met 500 mg tweemaal daags met tussenpozen van twee tot vier weken; bij kinderen en adolescenten die minder dan 50 kg wegen: dosisverlaging mag niet hoger zijn dan 10 mg/kg tweemaal daags om de twee weken).
Nierfalen
Toediening van Matever aan patiënten met nierinsufficiëntie kan dosisaanpassing vereisen. Bij patiënten met een ernstige leverfunctiestoornis wordt aanbevolen de nierfunctie te evalueren alvorens de dosering vast te stellen (zie rubriek 4.2).
Zelfmoord
Gevallen van zelfmoord, zelfmoordpogingen, zelfmoordgedachten en zelfmoordgedrag zijn gemeld bij patiënten die werden behandeld met anti-epileptica (waaronder levetiracetam). Een meta-analyse van gerandomiseerde, placebogecontroleerde onderzoeken met anti-epileptica toonde een licht verhoogd risico op zelfmoordgedachten en zelfmoordgedrag. Het mechanisme van dit risico is niet bekend.
Bijgevolg moeten patiënten worden gecontroleerd op tekenen van depressie en/of zelfmoordgedachten en -gedrag, en moet een passende behandeling worden overwogen. Patiënten (en zorgverleners) moeten erop worden gewezen dat als er tekenen van depressie en/of zelfmoordgedachten of zelfmoordgedrag optreden, medische hulp moet worden gezocht.
Pediatrische populatie
Beschikbare gegevens bij kinderen duiden niet op een invloed op de groei en puberteit. De langetermijneffecten op leren, intelligentie, groei, endocriene functie, puberteit en reproductievermogen bij kinderen zijn echter niet bekend.
Hulpstoffen
Dit geneesmiddel bevat 2,49 mmol (of 57,21 mg) natrium per maximale enkelvoudige dosis (0,83 mmol (of 19,07 mg) natrium per 5 ml). Hiermee moet rekening worden gehouden bij patiënten die een natriumarm dieet volgen.
04.5 Interacties met andere geneesmiddelen en andere vormen van interactie
Anti-epileptica
Gegevens uit klinische premarketingonderzoeken bij volwassenen geven aan dat Matever geen invloed heeft op de serumconcentraties van bestaande anti-epileptica (fenytoïne, carbamazepine, valproïnezuur, fenobarbital, lamotrigine, gabapentine en primidon) en dat deze anti-epileptica de farmacokinetiek van Matever niet beïnvloeden.
Net als bij volwassenen zijn er bij pediatrische patiënten die doses levetiracetam tot 60 mg/kg/dag kregen toegediend, geen bewijs van klinisch significante interacties met andere geneesmiddelen.
Een retrospectieve evaluatie van farmacokinetische interacties bij kinderen en adolescenten met epilepsie (4 tot 17 jaar) bevestigde dat aanvullende therapie met oraal toegediend levetiracetam geen invloed had op de steady-state serumconcentraties van gelijktijdig toegediende carbamazepine en valproaat. Gegevens suggereerden echter een 20% hogere klaring van levetiracetam bij kinderen die enzyminducerende anti-epileptica gebruikten. Er is geen dosisaanpassing nodig.
probenecide
Van probenecide (viermaal daags 500 mg), een renale tubulaire secretieblokker, is aangetoond dat het de renale klaring van de primaire metaboliet remt, maar niet van levetiracetam. De concentratie van deze metaboliet blijft echter laag. Van andere geneesmiddelen die met actieve tubulaire secretie worden uitgescheiden, wordt verwacht dat ze de renale klaring van de metaboliet verminderen. Het effect van levetiracetam op probenecide is niet onderzocht en het effect van levetiracetam op andere actief uitgescheiden geneesmiddelen, bijv. NSAID's, sulfonamiden en methotrexaat zijn niet bekend.
Orale anticonceptiva en andere farmacokinetische interacties
Levetiracetam 1000 mg per dag had geen invloed op de farmacokinetiek van orale anticonceptiva (ethinylestradiol en levonorgestrel); de endocriene parameters (luteïniserend hormoon en progesteron) werden niet gewijzigd. Levetiracetam 2000 mg per dag had geen invloed op de farmacokinetiek van digoxine en warfarine; de protrombinetijden werden niet veranderd. Gelijktijdige toediening van digoxine, orale anticonceptiva en warfarine had geen invloed op de farmacokinetiek van levetiracetam.
Alcohol
Er zijn geen gegevens over de interactie van levetiracetam met alcohol
04.6 Zwangerschap en borstvoeding
Zwangerschap
Postmarketinggegevens van verschillende prospectieve zwangerschapsregisters hebben de resultaten gedocumenteerd van blootstelling aan levetiracetam monotherapie bij meer dan 1000 vrouwen tijdens het eerste trimester van de zwangerschap. Over het algemeen duiden deze gegevens niet op een substantiële toename van het risico op ernstige aangeboren afwijkingen, hoewel een teratogeen risico niet volledig kan worden uitgesloten. Therapie met meerdere anti-epileptica gaat gepaard met een hoger risico op aangeboren afwijkingen dan monotherapie en daarom moet monotherapie worden overwogen. Dierstudies hebben reproductietoxiciteit aangetoond (zie rubriek 5.3).
Matever wordt niet aanbevolen, tenzij klinisch noodzakelijk, tijdens de zwangerschap en bij vrouwen in de vruchtbare leeftijd die geen anticonceptiemethoden gebruiken.
Net als bij andere anti-epileptica kunnen fysiologische veranderingen die samenhangen met zwangerschap de plasmaconcentraties van levetiracetam beïnvloeden. Tijdens de zwangerschap werden verlaagde plasmaconcentraties van levetiracetam waargenomen. Deze afname is het meest uitgesproken tijdens het derde trimester (tot 60% van de uitgangsconcentratie vóór de zwangerschap). Zwangere vrouwen die met levetiracetam worden behandeld, moeten vanuit klinisch oogpunt zorgvuldig worden gevolgd. Stopzetting van anti-epileptische behandelingen kan leiden tot een verergering van de ziekte die schadelijk kan zijn voor de moeder en de foetus.
Voedertijd
Levetiracetam wordt uitgescheiden in de moedermelk. Daarom wordt borstvoeding niet aanbevolen.Als behandeling met levetiracetam echter noodzakelijk wordt tijdens de borstvoeding, moet de baten/risicoverhouding van de behandeling worden afgewogen, rekening houdend met het belang van borstvoeding.
Vruchtbaarheid
In dierstudies werd geen effect op de vruchtbaarheid gevonden (zie rubriek 5.3). Er zijn geen klinische gegevens beschikbaar; het potentiële risico bij mensen is niet bekend.
04.7 Beïnvloeding van de rijvaardigheid en het vermogen om machines te bedienen
Er is geen onderzoek gedaan naar de rijvaardigheid en het vermogen om machines te bedienen.
Gezien de mogelijk verschillende individuele gevoeligheid, kunnen sommige patiënten slaperigheid of andere symptomen ervaren die verband houden met de werking op het centrale zenuwstelsel, vooral aan het begin van de behandeling of na een verhoging van de dosis. Daarom is voorzichtigheid geboden bij patiënten die bezig zijn met activiteiten die een hoge concentratie vereisen, zoals het besturen van voertuigen of het bedienen van machines. Patiënten dienen te worden geadviseerd niet te rijden of machines te bedienen totdat is vastgesteld dat hun vermogen om deze activiteiten uit te voeren niet wordt beïnvloed.
04.8 Bijwerkingen
Samenvatting van het veiligheidsprofiel
Het hieronder weergegeven bijwerkingenprofiel is gebaseerd op de analyse van gepoolde placebogecontroleerde klinische onderzoeken voor alle onderzochte indicaties voor in totaal 3416 patiënten die met levetiracetam werden behandeld. Deze gegevens worden aangevuld met het gebruik van levetiracetam in overeenkomstige open-label extensieonderzoeken, evenals vanaf postmarketingervaring De meest frequent gemelde bijwerkingen waren nasofaryngitis, slaperigheid, hoofdpijn, vermoeidheid en duizeligheid Het veiligheidsprofiel van levetiracetam is over het algemeen vergelijkbaar tussen leeftijdsgroepen (volwassen en pediatrische patiënten) en de goedgekeurde indicaties voor de behandeling van epilepsie Aangezien blootstelling aan intraveneuze levetiracetam beperkt is en de orale en intraveneuze formuleringen bio-equivalent zijn, is informatie over de veiligheid van intraveneuze levetiracetam geldig op basis van die van levetiracetam voor oraal gebruik.
Tabel met bijwerkingen
Bijwerkingen die zijn gemeld tijdens klinische onderzoeken (volwassenen, adolescenten, kinderen en zuigelingen ouder dan 1 maand) en uit postmarketingervaring, worden weergegeven in de volgende tabel per systeem/orgaanklasse en frequentie wordt als volgt gedefinieerd: zeer vaak (≥1 / 10); gemeenschappelijk (≥1 / 100,
Beschrijving van geselecteerde bijwerkingen
Het risico op anorexia is groter wanneer topiramaat gelijktijdig wordt toegediend met levetiracetam.
In talrijke gevallen van alopecia is genezing waargenomen na stopzetting van de behandeling met levetiracetam.
In sommige gevallen van pancytopenie werd beenmergsuppressie vastgesteld.
Pediatrische populatie
Bij patiënten in de leeftijd van 1 maand tot minder dan 4 jaar werden in totaal 190 patiënten behandeld met levetiracetam in placebogecontroleerde en open-label extensieonderzoeken. Zestig van deze patiënten werden behandeld met levetiracetam in placebogecontroleerde onderzoeken. Bij patiënten van 4 tot 16 jaar werden in totaal 645 patiënten behandeld met levetiracetam in placebogecontroleerde en open-label extensieonderzoeken. 233 van deze patiënten werden behandeld met levetiracetam in placebogecontroleerde onderzoeken. In beide pediatrische leeftijdsgroepen zijn deze gegevens geïntegreerd met postmarketingervaring met het gebruik van levetiracetam.
Het bijwerkingenprofiel van levetiracetam is over het algemeen vergelijkbaar tussen leeftijdsgroepen en tussen de goedgekeurde epilepsie-indicaties. In placebogecontroleerde klinische onderzoeken waren de veiligheidsresultaten bij pediatrische patiënten consistent met het veiligheidsprofiel van levetiracetam bij volwassenen, met uitzondering van gedrags- en psychiatrische bijwerkingen die vaker voorkwamen bij kinderen dan bij volwassenen.Bij kinderen en adolescenten van 4-16 jaar werden braken (zeer vaak, 11,2%), agitatie (vaak, 3,4%) vaker gemeld dan in andere leeftijdsgroepen of in het algemene veiligheidsprofiel.), stemmingswisselingen (vaak, 2,1 %), affectieve labiliteit (vaak, 1,7%), agressie (vaak, 8,2%), abnormaal gedrag (vaak, 5,6%) en lethargie (vaak, 3,9%) Bij zuigelingen en kinderen van 1 maand tot jonger dan 4 jaar, prikkelbaarheid werd vaker gemeld dan in andere leeftijdsgroepen of in het algemene veiligheidsprofiel (zeer vaak, 11,7%) en abnormale coördinatie (vaak, 3,3%).
Een veiligheidsonderzoek bij pediatrische patiënten, uitgevoerd volgens een niet-inferioriteit, dubbelblind, placebogecontroleerd ontwerp, evalueerde de cognitieve en neuropsychologische effecten van levetiracetam bij kinderen van 4 tot 16 jaar met partieel beginnende aanvallen. Levetiracetam bleek niet anders (niet inferieur) te zijn aan placebo in de verandering ten opzichte van baseline in de score verkregen in de subtest "Attentie en geheugen" van de Leiter-R-schaal (MemoryScreenSamengestelde scoreDe resultaten met betrekking tot gedrags- en emotionele functies wezen op een verslechtering, bij patiënten die met levetiracetam werden behandeld, van agressief gedrag, gemeten op een gestandaardiseerde en systematische manier, met behulp van een gevalideerd hulpmiddel (CBCL - Achenbach Checklist voor kindergedrag). Echter, proefpersonen die levetiracetam innamen in het open-label lange termijn follow-up onderzoek, ondervonden gemiddeld geen verslechtering van hun gedrags- en emotionele functies; in het bijzonder verslechterden de beoordelingen van agressie in gedrag niet in vergelijking met baseline.
Melding van vermoedelijke bijwerkingen
Het melden van vermoedelijke bijwerkingen die optreden na toelating van het geneesmiddel is belangrijk omdat het een continue controle van de baten/risicoverhouding van het geneesmiddel mogelijk maakt. Beroepsbeoefenaren in de gezondheidszorg wordt verzocht alle vermoedelijke bijwerkingen te melden via het nationale meldsysteem. in "Bijlage V .
04.9 Overdosering
Symptomen
Slaperigheid, agitatie, agressie, verminderd bewustzijn, ademhalingsdepressie en coma zijn waargenomen bij overdoses met Matever.
Behandeling van een overdosis
Er is geen specifiek antidotum voor levetiracetam. Behandeling van overdosering met levetiracetam dient symptomatisch te zijn en kan hemodialyse omvatten.De extractie-efficiëntie door dialyse is 60% voor levetiracetam en 74% voor de primaire metaboliet.
05.0 FARMACOLOGISCHE EIGENSCHAPPEN
05.1 Farmacodynamische eigenschappen
Farmacotherapeutische categorie: anti-epileptica, overige anti-epileptica.
ATC-code: N03AX14.
De werkzame stof, levetiracetam, is een pyrrolidon-derivaat (S-enantiomeer van? - ethyl-2-oxo-1-pyrrolidine-aceetamide), chemisch niet verwant aan bestaande anti-epileptica.
Werkingsmechanisme
Het werkingsmechanisme van levetiracetam is nog niet volledig verklaard, maar het lijkt anders te zijn dan de mechanismen van de huidige anti-epileptica. in vitro en in vivo suggereren dat levetiracetam de fundamentele cellulaire kenmerken en normale neurotransmissie niet verandert.
Opleiding in vitro tonen aan dat levetiracetam inwerkt op de intraneuronale Ca2+-spiegels door gedeeltelijk N-type Ca2+-stromen te remmen en door de afgifte van Ca2+ uit intraneuronale opslagplaatsen te verminderen. Bovendien keert het gedeeltelijk de reductie, veroorzaakt door zink en ?-Carboline, van de stromen veroorzaakt door GABA en glycine om. Opleiding in vitro ze ontdekten ook dat levetiracetam zich bindt aan een specifieke plaats in hersenweefsel van knaagdieren. Deze bindingsplaats is synaptisch vesikeleiwit 2A, waarvan wordt gedacht dat het betrokken is bij vesikelfusie en exocytose van neurotransmitters Levetiracetam en verwante analogen vertonen een mate van affiniteit voor binding aan synaptisch vesikeleiwit 2A die correleert met de potentie van hun anti-epileptische bescherming in de audiogene model van epilepsie bij muizen Deze bevinding suggereert dat de interactie tussen levetiracetam en synaptisch vesikeleiwit 2A een rol lijkt te spelen in het anti-epileptische werkingsmechanisme van het geneesmiddel.
Farmacodynamische effecten
Levetiracetam induceert een beschermende werking in een breed spectrum van diermodellen van partiële en primaire gegeneraliseerde epilepsie, zonder een pro-convulsief effect te hebben. De primaire metaboliet is inactief. Bij mensen is activiteit bij zowel partiële als gegeneraliseerde epilepsie-aandoeningen (epileptiforme afscheiding / fotoparoxysmale respons ) bevestigde het breedspectrum farmacologische profiel van levetiracetam.
Klinische werkzaamheid en veiligheid
Adjuvante therapie bij de behandeling van partieel beginnende aanvallen met of zonder secundaire generalisatie bij volwassenen, adolescenten en kinderen vanaf 4 jaar met epilepsie:
Bij volwassenen werd de werkzaamheid van levetiracetam aangetoond in 3 dubbelblinde, placebogecontroleerde onderzoeken met doses van 1000 mg, 2000 mg of 3000 mg/dag, verdeeld over 2 doses, voor een behandelingsduur van maximaal 18 weken. uitgebreide analyse was het percentage patiënten dat een vermindering van de frequentie van partiële aanvallen per week bereikte in de behandelingsperiode met stabiele dosis (12/14 weken), gelijk aan of groter dan 50% ten opzichte van baseline, 27,7%, 31,6% en 41,3% van de patiënten die werden behandeld met respectievelijk 1000, 2000 of 3000 mg levetiracetam, en 12,6% voor patiënten die werden behandeld met placebo.
Pediatrische populatie
De werkzaamheid van levetiracetam bij pediatrische patiënten (4 tot 16 jaar) werd aangetoond in een dubbelblind, placebogecontroleerd onderzoek met 198 patiënten en een behandelingsduur van 14 weken. dosis van 60 mg/kg/dag (tweemaal daags).
44,6% van de met levetiracetam behandelde patiënten en 19,6% van de met placebo behandelde patiënten had een vermindering van 50% of meer in de frequentie van partiële aanvallen per week vanaf baseline. Bij voortgezette langdurige behandeling was 11,4% van de patiënten ten minste 6 maanden aanvalsvrij en 7,2% was ten minste 1 jaar aanvalsvrij.
Monotherapie bij de behandeling van partieel beginnende aanvallen met of zonder secundaire generalisatie bij patiënten vanaf 16 jaar met nieuw gediagnosticeerde epilepsie.
De werkzaamheid van levetiracetam als monotherapie werd aangetoond in een dubbelblind vergelijkend non-inferioriteitsonderzoek met parallelle groepen versus carbamazepine (CR) met gecontroleerde afgifte bij 576 patiënten van 16 jaar of ouder met nieuwe of nieuwe epilepsie. alleen niet-uitgelokte partiële aanvallen of gegeneraliseerde tonisch-clonische aanvallen hebben Patiënten werden gerandomiseerd naar carbamazepine CR 400 - 1200 mg / dag of levetiracetam 1000 - 3000 mg / dag en de behandeling duurde tot 121 weken op basis van de respons.
Bij 73,0% van de met levetiracetam behandelde patiënten en bij 72,8% van de met carbamazepine CR behandelde patiënten werd gedurende een periode van 6 maanden vrij van aanvallen bereikt; het gecorrigeerde absolute verschil tussen behandelingen was 0,2% (95% BI: 7,8 - 8,2). Meer dan de helft van de proefpersonen bleef 12 maanden vrij van aanvallen (respectievelijk 56,6% en 58,5% van de proefpersonen die werden behandeld met levetiracetam en carbamazepine CR).
In een studie die de klinische praktijk weerspiegelt, kon gelijktijdige anti-epileptische behandeling worden stopgezet bij een beperkt aantal patiënten die reageerden op aanvullende therapie met levetiracetam (36 van de 69 volwassen patiënten).
Adjuvante therapie bij de behandeling van myoclonische aanvallen bij volwassenen en adolescenten vanaf 12 jaar met juveniele myoclonische epilepsie.
De werkzaamheid van levetiracetam werd aangetoond in een 16 weken durende, dubbelblinde, placebogecontroleerde studie bij patiënten van 12 jaar of ouder met idiopathische gegeneraliseerde epilepsie met myoclonische aanvallen bij verschillende syndromen.De meerderheid van de patiënten had juveniele myoclonische epilepsie.
In deze studie was de dosis levetiracetam 3000 mg/dag verdeeld over twee doses 58,3% van de met levetiracetam behandelde patiënten en 23,3% van de met placebo behandelde patiënten had een vermindering van ten minste 50% van het aantal dagen met myoclonische aanvallen per week. Na voortgezette langdurige behandeling was 28,6% van de patiënten gedurende ten minste 6 maanden vrij van myoclonische aanvallen en was 21,0% van de patiënten gedurende ten minste 1 jaar vrij van myoclonische aanvallen.
Adjuvante therapie bij de behandeling van primaire gegeneraliseerde tonisch-clonische aanvallen bij volwassenen en adolescenten vanaf 12 jaar met idiopathische gegeneraliseerde epilepsie.
De werkzaamheid van levetiracetam werd aangetoond in een dubbelblind, placebogecontroleerd onderzoek van 24 weken waarin volwassenen, adolescenten en een beperkt aantal kinderen met idiopathische gegeneraliseerde epilepsie met primaire gegeneraliseerde tonisch-klonische aanvallen (PGTC's) bij verschillende syndromen (juveniele myoclonische epilepsie, juveniele absentie-epilepsie, infantiele absentie-epilepsie of epilepsie met epileptische aanval van Great Male bij het ontwaken) In deze studie was de dosis levetiracetam 3000 mg/dag voor volwassenen en adolescenten of 60 mg/kg/dag voor kinderen, gegeven in twee verdeelde doses.
72,2% van de met levetiracetam behandelde patiënten en 45,2% van de met placebo behandelde patiënten hadden een vermindering van 50% of meer in de frequentie van PGTC-aanvallen per week. Na voortgezette langdurige behandeling was 47,4% van de patiënten gedurende ten minste 6 maanden vrij van tonisch-clonische aanvallen en was 31,5% gedurende ten minste één jaar vrij van tonisch-clonische aanvallen.
05.2 Farmacokinetische eigenschappen
Het farmacokinetische profiel werd beschreven na orale toediening. Een enkele dosis van 1500 mg levetiracetam, verdund in 100 ml van een compatibel verdunningsmiddel en intraveneus toegediend gedurende 15 minuten, is bio-equivalent aan 1500 mg oraal ingenomen levetiracetam in de vorm van drie tabletten van 500 mg.
De intraveneuze toediening van doses tot 4000 mg verdund in 100 ml van een 0,9% natriumchloride-oplossing toegediend via een infusie van 15 minuten en van doses tot 2500 mg verdund in 100 ml van een gelegeerde oplossing is geëvalueerd. minuten. De farmacokinetische en veiligheidsprofielen identificeerden geen veiligheidsimplicaties.
Levetiracetam is een zeer oplosbare en permeabele verbinding. Het farmacokinetische profiel is lineair met weinig intra- en interindividuele variabiliteit. Er is geen verandering in de klaring na herhaalde toediening Het tijdonafhankelijke farmacokinetische profiel van levetiracetam werd bevestigd na intraveneuze infusie van 1500 mg gedurende 4 dagen met een tweemaal daags
Er zijn geen aanwijzingen voor relevante circadiane, geslachts- en rasvariabiliteit Het farmacokinetische profiel is vergelijkbaar bij gezonde vrijwilligers en patiënten met epilepsie.
Volwassenen en adolescenten
Verdeling
De piekplasmaconcentratie (Cmax) die werd waargenomen bij 17 proefpersonen na een enkelvoudige intraveneuze dosis van 1500 mg per infuus gedurende 15 minuten was 51 ± 19 g/ml (rekenkundig gemiddelde ± standaarddeviatie).
Er zijn geen gegevens over weefseldistributie bij mensen.
Noch levetiracetam, noch zijn primaire metaboliet bindt significant aan plasma-eiwitten (
Het distributievolume van levetiracetam is ongeveer 0,5 tot 0,7 l/kg en ligt dicht bij het totale lichaamsvolume aan water.
Biotransformatie
Levetiracetam wordt niet uitgebreid gemetaboliseerd bij de mens.De belangrijkste metabole route (24% van de dosis) is de enzymatische hydrolyse van de acetamidegroep. De productie van de primaire metaboliet, ucb L057, wordt niet ondersteund door hepatische cytochroom P450-isovormen. Hydrolyse van de acetamidegroep is meetbaar in tal van weefsels, waaronder bloedcellen.De metaboliet ucb L057 is farmacologisch inactief.
Er werden ook twee minder belangrijke metabolieten geïdentificeerd. De ene werd verkregen door de hydroxylering van de pyrrolidonring (1,6% van de dosis) en de andere door de opening van de pyrrolidonring (0,9% van de dosis).
Andere onbekende componenten waren goed voor slechts 0,6% van de dosis.
In vivo er was geen bewijs van enantiomere interconversie voor levetiracetam of zijn primaire metaboliet.
In vitro, is aangetoond dat levetiracetam en zijn primaire metaboliet de activiteiten van de belangrijkste humane levercytochroom P450-isovormen (CYP3A4, 2A6, 2C9, 2C19, 2D6, 2E1 en 1A2) niet remmen,
glucuronyltransferase (UGT1A1 en UGT1A6) en epoxidehydroxylase Bovendien heeft levetiracetam geen invloed op de glucuronidering in vitro van valproïnezuur.
In humane hepatocytenculturen had levetiracetam weinig of geen effect op CYP1A2, SULT1E1 of UGT1A1. Levetiracetam veroorzaakte matige inductie van CYP2B6 en CYP3A4. De gegevens in vitro en de gegevens in vivo gerelateerd aan de interactie met orale anticonceptiva, digoxine en warfarine, geven aan dat er geen significante enzyminductie wordt verwacht in vivo. Vandaar de interactie van Matever met andere stoffen, of andersom, het is onwaarschijnlijk.
Eliminatie
De plasmahalfwaardetijd bij volwassenen is 7 ± 1 uur en verandert niet met de dosis, de toedieningsweg of herhaalde toediening De gemiddelde totale lichaamsklaring is 0,96 ml/min/kg.
De belangrijkste uitscheidingsroute is de urineweg, die gemiddeld verantwoordelijk is voor de eliminatie van 95% van de toegediende dosis (ongeveer 93% van de dosis wordt uitgescheiden in 48 uur) Fecale eliminatie is goed voor slechts 0,3% van de dosis.
De cumulatieve uitscheiding via de urine van levetiracetam en zijn primaire metaboliet is verantwoordelijk voor de eliminatie van respectievelijk 66% en 24% van de dosis gedurende de eerste 48 uur.
De renale klaring van levetiracetam en ucb L057 is respectievelijk 0,6 en 4,2 ml/min/kg, wat aangeeft dat levetiracetam wordt uitgescheiden door glomerulaire filtratie met daaropvolgende tubulaire reabsorptie en dat de primaire metaboliet ook wordt uitgescheiden door actieve tubulaire secretie voorbij dan door glomerulaire filtratie. Eliminatie van levetiracetam houdt verband met de creatinineklaring.
Bejaarden
Bij de "ouderen" nam de halfwaardetijd toe met ongeveer 40% (van 10 naar 11 uur). Dit komt door een verminderde nierfunctie bij deze populatie (zie rubriek 4.2).
Nierfunctiestoornis
De schijnbare lichaamsklaring van zowel levetiracetam als zijn primaire metaboliet correleert met de creatinineklaring. Bij patiënten met een matige tot ernstige nierfunctiestoornis wordt daarom aanbevolen om de dagelijkse onderhoudsdosering van Matever aan te passen op basis van de creatinineklaring (zie rubriek 4.2).
Bij volwassen patiënten met anurische nierinsufficiëntie in het eindstadium was de halfwaardetijd respectievelijk ongeveer 25 en 3,1 uur in de interdialyse- en tijdens dialyseperioden.
De fractie verwijderd levetiracetam was 51% tijdens een typische 4-uurs dialyse.
leverfunctiestoornis
Bij proefpersonen met een lichte tot matige leverfunctiestoornis was er geen significante verandering in de klaring van levetiracetam. Bij de meerderheid van de proefpersonen met een ernstige leverfunctiestoornis was de klaring van levetiracetam met meer dan 50% verminderd als gevolg van een gelijktijdige nierfunctiestoornis (zie rubriek 4.2).
Pediatrische populatie
Kinderen (van 4 tot 12 jaar)
Farmacokinetische onderzoeken bij pediatrische patiënten zijn niet uitgevoerd na intraveneuze toediening. Echter, op basis van de farmacokinetische kenmerken van levetiracetam, de farmacokinetiek bij volwassenen na intraveneuze toediening en de farmacokinetiek bij kinderen na orale toediening, is de verwachte blootstelling (AUC) aan levetiracetam bij pediatrische patiënten van 4 tot 12 jaar vergelijkbaar na intraveneuze en orale toediening.
Na een enkelvoudige orale toediening (20 mg/kg) bij kinderen (6 tot 12 jaar) met epilepsie was de halfwaardetijd van levetiracetam 6 uur. De schijnbare, voor het lichaamsgewicht gecorrigeerde klaring was ongeveer hoger 30% in vergelijking met volwassenen met epilepsie .
Na herhaalde orale toediening (20 tot 60 mg/kg/dag) aan epileptische kinderen (4 tot 12 jaar) werd levetiracetam snel geabsorbeerd. Piekplasmaconcentraties werden 0,5 tot 1,0 uur na toediening waargenomen. Lineaire en dosisproportionele verhogingen werden waargenomen voor piekplasmaconcentraties en oppervlakte onder de curve.De eliminatiehalfwaardetijd was ongeveer 5 uur. De schijnbare lichaamsklaring was 1,1 ml/min/kg.
05.3 Gegevens uit het preklinisch veiligheidsonderzoek
Niet-klinische gegevens duiden niet op een risico voor mensen. Deze gegevens zijn afkomstig van conventioneel onderzoek op het gebied van veiligheidsfarmacologie, genotoxiciteit en carcinogeen potentieel.
Bijwerkingen die niet zijn waargenomen in klinische onderzoeken, maar die zijn waargenomen bij ratten en in mindere mate bij muizen, bij blootstellingsniveaus die vergelijkbaar zijn met blootstellingsniveaus bij de mens en met mogelijke relevantie voor klinisch gebruik, waren de indexen van de respons van leververanderingen, zoals gewichtstoename en centrilobulaire hypertrofie, vetinfiltratie en verhoging van leverenzymen in plasma.
Er werden geen nadelige effecten op de mannelijke en vrouwelijke vruchtbaarheid of reproductieve capaciteit waargenomen bij ratten bij doses tot 1800 mg/kg/dag (6 maal de MRHD (Maximaal aanbevolen dagelijkse dosis voor de mens) op basis van mg/m2 of op basis van blootstelling), zowel in de oudergeneratie als in de F1-generatie.
Twee embryo-foetale ontwikkelingsstudies (EFD: Embryo-foetale ontwikkeling) werden uitgevoerd bij ratten met 400, 1200 en 3600 mg/kg/dag. Bij 3600 mg / kg / dag was er in slechts één van de 2 EFD-onderzoeken een lichte afname van het foetale gewicht geassocieerd met een marginale toename van skeletveranderingen / kleine anomalieën. Er was geen effect op de embryonale mortaliteit en er was ook geen toename van de incidentie van misvormingen.Geen waargenomen schadelijk effectniveau) was 3600 mg/kg/dag voor drachtige vrouwelijke ratten (12 maal de maximaal aanbevolen dagelijkse dosis voor de mens (MRHD) op basis van mg/m2) en 1200 mg/kg/dag voor foetussen.
Vier embryo-foetale ontwikkelingsstudies werden uitgevoerd bij konijnen met doses van 200, 600, 800, 1200 en 1800 mg/kg/dag. De dosis van 1800 mg/kg/dag veroorzaakte duidelijke maternale toxiciteit en een verlaagd foetaal gewicht in combinatie met een hogere incidentie van foetussen met cardiovasculaire/skeletafwijkingen. De NOAEL was 2).
Een peri- en postnatale ontwikkelingsstudie werd uitgevoerd bij ratten met doses levetiracetam van 70, 350, 1800 mg/kg/dag. De NOAEL was ≥ 1800 mg/kg/dag voor F0 vrouwtjes en voor de F1-generatie voor overleving, groei en ontwikkeling tot aan het spenen (6 keer de MRHD op basis van mg/m2).
Studies bij ratten en honden, bij pasgeboren en juveniele dieren, hebben aangetoond dat er geen nadelige effecten optreden in een van de standaard ontwikkelings- of rijpingseindpunten bij doses tot 1.800 mg/kg/dag (6-17 maal de MRHD op basis van mg/m2 ).
Milieurisicobeoordeling (Milieurisicobeoordeling, WAS)
Het is onwaarschijnlijk dat het gebruik van Matever in overeenstemming met de informatie in de Samenvatting van de Productkenmerken resulteert in een onaanvaardbare impact op het milieu (zie rubriek 6.6).
06.0 FARMACEUTISCHE INFORMATIE
06.1 Hulpstoffen
Natriumacetotrihydraat
Ijsazijn
Natriumchloride
Water voor injecties
06.2 Incompatibiliteit
Dit geneesmiddel mag niet gemengd worden met andere geneesmiddelen dan die vermeld in rubriek 6.6.
06.3 Geldigheidsduur
2 jaar.
Vanuit microbiologisch oogpunt moet het product onmiddellijk na verdunning worden gebruikt. Indien het niet onmiddellijk wordt gebruikt, zijn de bewaartijden en -omstandigheden voorafgaand aan gebruik de verantwoordelijkheid van de gebruiker en zouden deze normaal niet langer zijn dan 24 uur bij 2 tot 8 ° C, tenzij verdunning is uitgevoerd onder gecontroleerde en gevalideerde aseptische omstandigheden.
06.4 Speciale voorzorgsmaatregelen bij bewaren
Voor dit geneesmiddel zijn er geen speciale bewaarcondities.
Voor de bewaarcondities van het verdunde geneesmiddel, zie rubriek 6.3.
06.5 Aard van de primaire verpakking en inhoud van de verpakking
Glazen injectieflacon van 5 ml (Type I) met broombutylrubberen stoppen en verzegeld met een aluminium flip-off dop.
Elke doos bevat 10 injectieflacons.
06.6 Instructies voor gebruik en verwerking
Zie Tabel 1 voor de aanbevolen bereiding en toediening van Matever-concentraat om een totale dagelijkse dosis van 500 mg, 1000 mg, 2000 mg of 3000 mg te verkrijgen, verdeeld over twee doses.
Tabel 1. Bereiding en toediening van Matever-concentraat.
Dit geneesmiddel is uitsluitend bedoeld voor eenmalig gebruik en alle ongebruikte oplossing moet worden weggegooid.
Matever-concentraat bleek fysiek compatibel en chemisch stabiel te zijn gedurende ten minste 24 uur wanneer het werd gemengd met de volgende verdunningsmiddelen en bewaard in PVC-zakken bij een gecontroleerde kamertemperatuur van 15 - 25 ° C.
Verdunners:
• Natriumchloride (0,9%) voor injectie
• Ringer's lactaatinjectie
• Dextrose 5% injecteerbaar preparaat
Geneesmiddelen die deeltjes of troebelheid bevatten, mogen niet worden gebruikt.
Ongebruikte medicijnen en afval afkomstig van dit medicijn moeten worden weggegooid in overeenstemming met de lokale regelgeving.
07.0 HOUDER VAN DE VERGUNNING VOOR HET IN DE HANDEL BRENGEN
Pharmathen SA 6, Dervenakion str., 153 51 Pallini Attiki, Griekenland
08.0 NUMMER VAN DE VERGUNNING VOOR HET IN DE HANDEL BRENGEN
EU / 1/11/711/030
041466309
09.0 DATUM VAN EERSTE VERGUNNING OF VERLENGING VAN DE VERGUNNING
Datum eerste autorisatie: 03 oktober 2011
10.0 DATUM VAN HERZIENING VAN DE TEKST
D.CCE januari 2015





















-cos-cause-e-rimedi.jpg)

